Què és l’Anèmia de Fanconi i a qui afecta?
L’Anèmia de Fanconi és una malaltia hereditària de molt baixa freqüència entre la població. Malgrat el seu nom, acostuma a caracteritzar-per moltes altres manifestacions a part de l’anèmia.
Les més rellevants:
- Insuficiència medul·lar progressiva (anèmia, leucopènia i trombocitopènia).
- Malformacions congènites
- especialment a desenvolupar leucèmies mieloblàstiques agudes (LMA), síndromes mielodisplàstiques (SMD) i tumors sòlids.
 La seva freqüència és d’1 cada 300.000. Es tracta d’una malaltia genètica recessiva en la majoria dels casos. Per tant, perquè un individu pateixi aquesta malaltia, les dues còpies dels gens involucrats en l’Anèmia de Fanconi (l’heretada del pare i l’heretada de la mare) han d’estar afectades. L’afecció generalment es diagnostica en nens entre els 3 i 14 anys d’edat. Quan els gens estan implicats en una predisposició tumoral en portadors, és important realitzar un seguiment adequat en unitats especialitzades. És fonamental rebre un adequat consell genètic dels familiars en risc, especialment en parelles portadores.
La seva freqüència és d’1 cada 300.000. Es tracta d’una malaltia genètica recessiva en la majoria dels casos. Per tant, perquè un individu pateixi aquesta malaltia, les dues còpies dels gens involucrats en l’Anèmia de Fanconi (l’heretada del pare i l’heretada de la mare) han d’estar afectades. L’afecció generalment es diagnostica en nens entre els 3 i 14 anys d’edat. Quan els gens estan implicats en una predisposició tumoral en portadors, és important realitzar un seguiment adequat en unitats especialitzades. És fonamental rebre un adequat consell genètic dels familiars en risc, especialment en parelles portadores.
Consell genètic
En tractar-se en la majoria dels casos de mutacions recessives, perquè un individu pateixi Anèmia de Fanconi, els dos al·lels d’un determinat gen FANC heretats dels seus pares han de tenir la mutació patogènica.
Si un home i una dona, ambdós portadors de mutacions en un determinat gen FANC tenen descendència, 3 de cada 4 fills, de mitjana, seran sans (de mitjana, 2 seran portadors i 1 serà completament normal: no tindrà afectat cap dels dos al·lels d’aquest gen).
No obstant això, de mitjana, 1 de cada 4 fills tindrà mutacions en ambdós al·lels, per tant, patirà la malaltia amb una severitat difícil de definir con antelació. En el cas del gen FANCB, en estar en el cromosoma X, tots els afectats són homes i només les mares són portadores. En aquestes famílies és molt important detectar dones portadores en edat reproductiva degut a l’alt risc d’engendrar un fill baró afecte.
Els individus que tenen una còpia funcional i l’altra mutada es defineixen com a portadors de la malaltia. Tret del cas de portadors amb una mutació en els gens FANCD1/BRCA2, FANCS/BRCA1, FANCN/PALB2 o FANCJ/BRIP1, que afecten una minoria dels pacients, no existeix cap evidència que portadors d’AF tinguin riscos de patir malalties amb una freqüència més gran que la resta de la població.
Altres consideracions dins del consell genèric (planificació familiar, diagnòstic prenatal o diagnòstic preimplantacional):
És important contactar amb especialistes per rebre consell genètic que ajudarà una persona o una família a entendre el risc de patir una malaltia genètica, educar les persones sobre aquesta malaltia i avaluar el risc de transmetre la malaltia als seus fills, per tenir una panificació familiar adequada i un diagnòstic prenatal o preimplantacional.
Freqüentment l’assessor genètic treballa amb familiars per identificar les persones en risc. En cas de ser apropiat o necessari, parlarà de tests genètics, interpretarà resultats i supervisarà totes les anàlisis addicionals, ajudes o opcions d’investigació a l’abast dels membres de la família. Els assessors genètics treballen com a part d’un equip de salut, juntament amb metges especialitzats, treballadors socials, personal d’infermeria, genetistes clínics i altres especialistes per ajudar les famílies a prendre decisions informades sobre la seva salut. També seran font d’ajuda i suport en moments difícils
Si desitgeu contactar amb els especialistes que us poden ajudar en el diagnòstic, seguiment i tractament de pacients amb anèmia de Fanconi podeu fer-ho de la manera següent:
Fundación Anemia de Fanconi España
secretariaFAF@anemiadefanconi.org
654 617 808
Quines són les causes de l’Anèmia de Fanconi?
 L’Anèmia de Fanconi és una malaltia complexa des del punt de vista genètic, ja que fins el moment s’han descrit 22 gens involucrats en aquesta malaltia. La manca de funció de qualsevol d’aquests 22 gens produeix la malaltia. Aquests gens anormals danyen les cèl·lules, la qual cosa impedeix reparar l’ADN danyat. Aquesta malaltia es deu principalment a mutacions en els gens; existeixen algunes correlacions entre subtipus d’Anèmia de Fanconi, mutacions genètiques i el comportament de la malaltia.
L’Anèmia de Fanconi és una malaltia complexa des del punt de vista genètic, ja que fins el moment s’han descrit 22 gens involucrats en aquesta malaltia. La manca de funció de qualsevol d’aquests 22 gens produeix la malaltia. Aquests gens anormals danyen les cèl·lules, la qual cosa impedeix reparar l’ADN danyat. Aquesta malaltia es deu principalment a mutacions en els gens; existeixen algunes correlacions entre subtipus d’Anèmia de Fanconi, mutacions genètiques i el comportament de la malaltia.
En els pacients amb mutacions de FANC C l’aplàsia és més precoç i la supervivència més curta que en els portadors de mutacions en FANC A i FANC G.
Els pacients amb FANC G tenen més risc de patir leucèmies i tumors sòlids que els del tipus FANC A.
Els subtipus FANC D1 i FANC N comporten risc de desenvolupar medul·loblastomes o tumors de Wilms. Les mutacions heterozigotes en FANC D1, FANC J i FANC N comporten un risc incrementat de càncer de mama.
Quins són els símptomes de l’Anèmia de Fanconi?
Aproximadament el 90 % dels nens i nenes amb Anèmia de Fanconi tenen alteracions de la funció de la medul·la òssia que condueixen a una anèmia aplàstica. Els individus afectats pateixen cansament i debilitat degut a l’anèmia, infeccions degut a la neutropènia i problemes de coagulació degut a la trombocitopènia.
Les característiques clíniques associades a l’Anèmia de Fanconi són el retard del creixement pre i postnatal, malformacions renals, gastrointestinals, genitourinàries, cardíaques i esquelètiques, cap, ulls i boca petits, hipogonadisme, sordesa parcial, anormalitats cutànies com ara híper o hipopigmentació i taques “cafè amb llet” i elevats nivells d’a-fetoproteïna en sang. Tres de cada deu pacients, no obstant això, no presenten cap malformació.
Els problemes hematològics acostumen a aparèixer en l’edat escolar, al voltant dels 7 anys, encara que això és molt variable. Les alteracions hematològiques afecten a la majoria dels pacients d’Anèmia de Fanconi abans dels 40 anys. El 90 % dels casos es diagnostica abans de l’adolescència. Els pacients mostren recomptes anormalment baixos de cèl·lules sanguínies, tant de glòbuls vermells (anèmia), glòbuls blancs (leucopènia) com de plaquetes (trombopènia). La primera manifestació acostuma a ser una trombopènia aïllada en més de la meitat dels casos; s’observen petèquies o hematomes, o episodis de sagnat nasal o gastrointestinal. Posteriorment es fan evidents els signes d’anèmia que inclouen principalment pal·lidesa, astènia i anorèxia. La tendència a patir processos infecciosos (secundaria a la deficiència de glòbuls blancs) acostuma a ser d’aparició tardana. Una vegada iniciada l’afectació hematològica, l’evolució acostuma a conduir a la pancitopènia (anèmia + leucopènia + trombopènia) en un període de temps extraordinàriament variable.
Com es diagnostica l’Anèmia de Fanconi?
- Diagnòstic clínic
El diagnòstic basat en observacions clíniques pot resultar extremadament dificultós degut a la gran variabilitat de símptomes que mostren els pacients d’Anèmia de Fanconi. Aquest fet, unit al fet que un 30 % dels pacients AF no mostra cap malformació, suposa que molts pacients d’Anèmia de Fanconi només vagin al metge quan els símptomes de la malaltia hematològica es presenten. La marcada reducció d’una o més sèries sanguínies acostuma a posar l’especialista davant la sospita d’una anèmia hereditària, que s’ha de confirmar mitjançant estudis clínics addicionals, així com amb anàlisis citogenètiques i moleculars.
- Diagnòstic genètic
L’Anèmia de Fanconi és una malaltia complexa des del punt de vista genètic, ja que fins al moment s’han descrit 22 gens involucrats en aquesta malaltia: FANCA, FANCB, FANCC, FANCD1/BRCA2, FANCD2, FANCE, FANCF, FANCG/XRCC9, FANCI, FANCJ/BRIP1/BACH1, FANCL, FANCM, FANCN/PALB2, FANCO/RAD51C, FANCP/SLX4/BTBD12, FANCQ/ERCC4/XPF, FANCR/RAD51, FANCS/BRCA1, FANCT/UBE2T, FANCU/XRCC2, FANCV/REV7,FANCW/RFWD3
La cromosòmica (ruptures de cromosomes) induïdes per agents que generen enllaços creuats en les cadenes d’ADN. Aquests estudis acostumen a realitzar-se sobre limfòcits de sang perifèrica o fibroblasts de pell. Aquests assajos s’han de realitzar per laboratoris experimentats en el reconeixement sistemàtic de malalties genètiques. La Xarxa Nacional d’Investigació en Anèmia de Fanconi ofereix als pacients espanyols la realització de confirmacions diagnòstiques de la malaltia, així com entrenament a laboratoris que vulguin especialitzar-ser en aquests assajos. En aproximadament un 20 % dels pacients d’Anèmia de Fanconi, la confirmació diagnòstica de la malaltia pot ser particularment complexa degut a un fenomen conegut com a mosaïcisme somàtic. El mosaïcisme somàtic deriva del fet que algunes cèl·lules de la sang poden corregir, per diferents mecanismes, la mutació del gen causant de la malaltia; per la qual cosa, en la sang del pacient cohabiten cèl·lules d’Anèmia de Fanconi i cèl·lules sanes.
El diagnòstic del subtipus genètic es realitza amb tècniques de seqüenciació (Sanger o NGS) i MLPA. Mitjançant l’estudi mutacional es poden identificar portadors de la malaltia i realitzar estudis de diagnòstic prenatal o preimplantacional.
- Diagnòstic diferencial
És important que l’equip tingui experiència en el control dels pacients d’Anèmia de Fanconi. Hi ha algunes malalties que es poden confondre en el diagnòstic. A tenir en compte:
-
- Disqueratosi congènita
- Anèmia de Blacfan-Diamond
- Síndrome de Shwachman-Diamond
- Neutropènia congènita severa
- Síndrome TAR (trombocitopènia amb absència de radis)
- Trombocitopènia amegacariocítica
- Síndrome de Baller-Gerold
- Síndrome de ruptura de Hijmegen
- Síndrome de Rothmund-Thomson
- Síndrome de Roberts
- Síndrome de ruptura de Varsòvia
- Focomèlia DK
- Síndrome de hidrocefàlia VACTERL
- Síndrome de Wiskott-ALdrich
Quin és el tractament de l'Anèmia de Fanconi?
Les transfusions d’hematies i plaquetes són necessàries quan l’anèmia i la trombocitopènia són intenses i simptomàtiques..

Els derivats androgènics (oximetolona a dosis de 2-4 mg/kg/dia) poden disminuir el grau d’anèmia i estalviar transfusions, però s’ha de controlar l’aparició d’efectes secundaris (virilització, acceleració del creixement, disfunció hepàtica i tumors hepàtics). El seu ús ha de ser transitori a l’espera d’un trasplantament de progenitors hematopoètics (trasplantament de medul·la òssia).
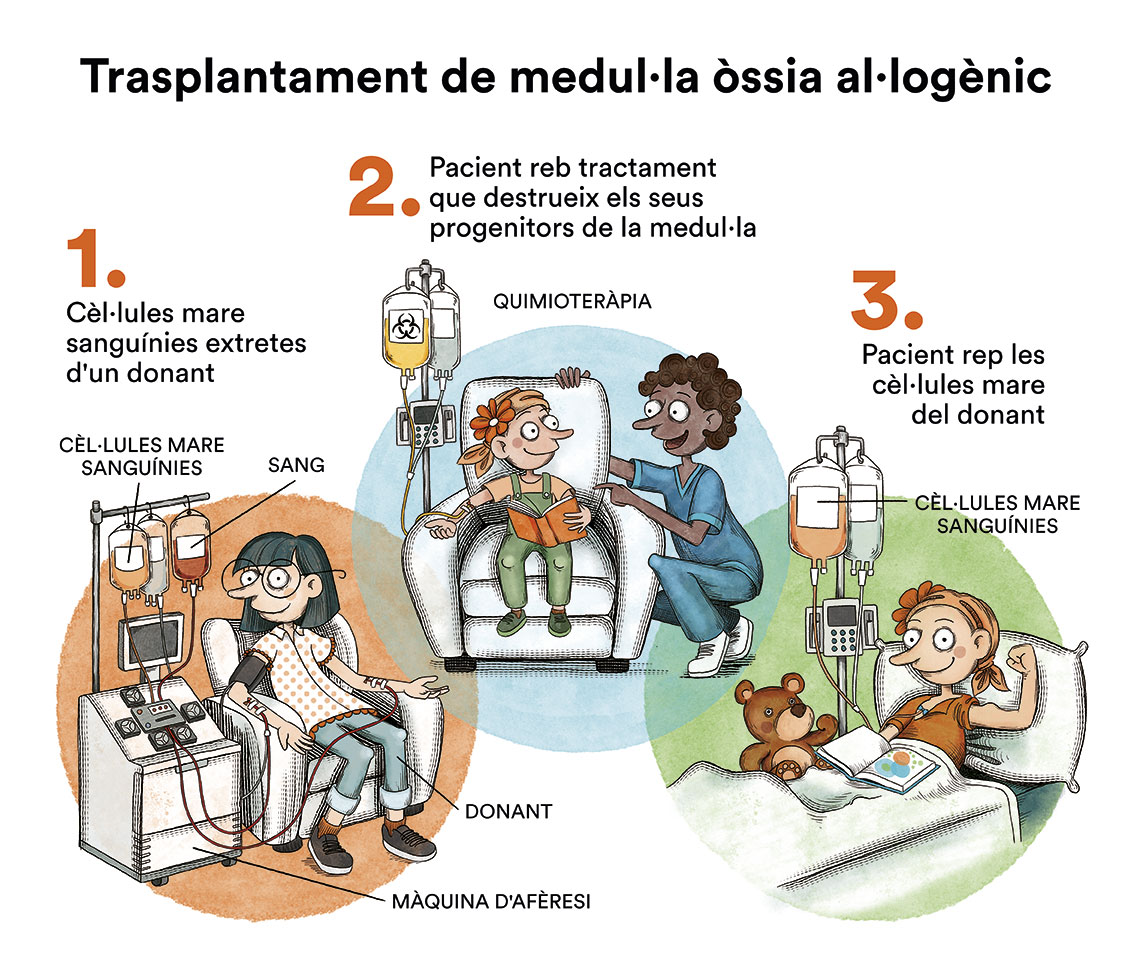
El trasplantament de progenitors hematopoètics és l’únic tractament que pot restaurar una hematopoesi normal, però no evita les lesions somàtiques ni prevé el desenvolupament de tumors sòlids. El donant ideal és un germà HLA idèntic però, actualment, si aquesta opció no existeix, els trasplantaments de donants voluntaris HLA compatibles també són molt bons.
L’ús dels clàssics règims mieloablatius amb ciclofosfamida i irradiació donen lloc a resultats dolents per la hipersensibilitat de les cèl·lules de l’Anèmia de Fanconi als agents genotòxics que produeixen greus lesions als teixits. Per això, s’han d’utilitzar règims de condicionament d’intensitat reduïda adaptats als pacients i evitar sempre la radioteràpia.
Els pacients amb Anèmia de Fanconi receptors d’un trasplantament de medul·la òssia requereixen un seguiment estricte pel risc de desenvolupar neoplàsies, que inclou revisions de creixement i endocrinològiques, citogenètiques en medul·la òssia i exàmens orals regulars amb biòpsia davant qualsevol lesió sospitosa.
Quines probabilitats tenen de curar-se els nens amb Anèmia de Fanconi?
 La probabilitat de transformació de la malaltia en una leucèmia mieloide aguda és del 30 % i la incidència de tumors sòlids del 28 % als 40 anys.
La probabilitat de transformació de la malaltia en una leucèmia mieloide aguda és del 30 % i la incidència de tumors sòlids del 28 % als 40 anys.
Sense trasplantament de medul·la òssia, la mitjana de supervivència dels pacients amb Anèmia de Fanconi és de 24 anys. La incidència d’insuficiència medul·lar arriba a ser del 90 % als 40 anys. La mort, en el 90 % dels casos, es deu a les alteracions hematològiques (aplasia medul·lar, síndromes mielodisplàsiques o leucèmia mieloide aguda) o a complicacions derivades del seu tractament.
Els trasplantaments realitzats a partir d’un germà compatible i sa ofereixen una probabilitat de supervivència lliure de malaltia hematològica de fins al 90 %. L’ús d’un donant HLA anònim no és òptim, però millora cada dia. El pronòstic associat al trasplantament és millor si el pacient ha rebut poques transfusions, si el pacient és jove i si no ha desenvolupat un procés leucèmic. Per tant, en cas que el pacient tingui un donant adequat, és recomanable realitzar el trasplantament abans que comenci a requerir suport transfusional, o quan els estudis citogenètics en la medul·la òssia suggereixin l’inici d’una mielodisplàsia. És per aquest motiu que és tan important un seguiment correcte d’aquests pacients a nivell de la seva sang perifèrica i medul·la òssia. La Red Nacional de Investigación en Anemia de Fanconi ha estandarditzat protocols per al trasplantament d’aquests pacients d’acord als millors estàndards internacionals, i ofereix informació als centres especialitzats que la sol·licitin. Contactar amb la Fundación Anemia de Fanconi.
Seguiment
- Seguiment hematològic
Els pacients amb Anèmia de Fanconi tenen un elevat risc de presentar anèmia aplàsica, síndromes mielodisplàsquies o leucèmia mieloide aguda. La probabilitat de transformació en LMA es del 30 % i la incidència de tumors sòlids del 28 % als 40 anys. És molt important realitzar un control rigorós dels seus paràmetres hematològics. A títol orientatiu, els controls recomanats són els següents:
-
- Hemograma mitjançant mostra de sang perifèrica cada 3-4 mesos
- Mielograma, citogenètica, anàlisis CD34+ i contingut CFCs) mitjançant aspirat de medul·la òssia cada 1 any-1,5 anys si no s’observen canvis significatius.
El seguiment hematològic del pacient ha de començar des del diagnòstic de la seva malaltia.
- Seguiment i prevenció de tumors de cap i coll
Els pacients amb Anèmia de Fanconi tenen una predisposició, molt més elevada que la resta de la població, a desenvolupar tumors sòlids. Per aquesta raó, el seguiment per part d’especialistes
que coneguin la malaltia és de vital importància. Hi ha un risc acumulat de patir algun tipus de tumor a 40 anys d’un 25 %. No hi ha casos descrits en pacients de menys de 10 anys. En pacients trasplantats el risc augmenta a partir del 5è any post-trasplantament. Les recomanacions per a revisions i seguiment rutinari són les següents:
-
- Pacients no trasplantats: des dels 10 anys d’edat
- Pacients trasplantats: des del moment del trasplantament independentment de la seva edat
S’ha d’establir vigilància de la cavitat bucal, nasofaringe, orofaringe, hipofaringe i laringe cada semestre si no hi ha canvis significatius. En cas que hi hagi canvis, s’ha de vigilar lichenplanus, leucoplàquia i eritroplàquia mitjançant una biòpsia cada 2-3 mesos. I si hi ha lesió sospitosa de carcinoma, prendre immediatament una biòpsia i realitzar seguiment cada 2-3 mesos, a més de realitzar una radiografia anual.
- Seguiment i prevenció de tumors ginecològics
Degut al risc de càncer de cèrvix, vagina i vulva, s’ha de començar amb el seguiment ginecològic a partir dels 16 anys o de la primera menstruació. De la mateixa manera, s’ha de realitzar un seguiment de control de mames també a partir de la primera menstruació o dels 20 anys.
El seguiment ha de consistir anualment en: un frotis Papanicolau, examen meticulós del tracto genital inferior, seguiment endocrí i test del virus del Papil·loma.
- Seguiment i cures dentals
Es recomana iniciar les revisions dentals a l’edat d’un any i mig i realitzar 2 revisions l’any. Es recomana posar en contacte el dentista que tracta un pacient d’Anèmia de Fanconi amb el seu metge especialista, amb l’objectiu que l’informi sobre les possibles complicacions associades a intervencions bucals.
Es recomana revisar lesions persistents, ulceracions sospitoses o leucoplàquies i posar-se en contacte amb l’especialista per si fos convenient realitzar una biòpsia del teixit bucal. Es recomana també posar-se en contacte amb l’especialista, en cas de sagnat continuat, o pèrdues dentals sense causa aparent.
En cas que el pacient presenti nivells anormalment baixos de plaquetes o glòbuls blancs, convé informar el dentista, per prevenir hemorràgies o infeccions en cas que el pacient s’hagi de sotmetre a una intervenció dental.
Enllaços d'interès sobre temes mèdics relacionats amb l’Anèmia de Fanconi
- Guia bàsica per al diagnòstic. Fundación Anemia de Fanconi España
- Seguiment de pacients d’Anèmia de Fanconi. Fundación Anemia de Fanconi España
Enllaços d'interès sobre altres temes relacionats amb l’Anèmia de Fanconi
MATERIALS LEUCÈMIA INFANTIL
- Els nadons també tenen leucèmia. Fundació Josep Carreras contra la leucèmia. (contingut en espanyol)
- Leucèmia infantil. Els petits imparables. Fundació Josep Carreras contra la leucèmia. (contingut en espanyol)
- Joc retallable Medulín. Fundació Josep Carreras contra la leucèmia. (contingut en espanyol)
La Fundació Josep Carreras disposa d’un conte “El nadó forçut” dirigit a nens o germans que pateixen leucèmia. Està especialment dirigit a nens fins als 6 anys. Si el vols demanar, pots enviar-nos un correu electrònic a imparables@fcarreras.es.
TRASPLANTAMENT DE MEDUL·LA ÒSSIA
- Guia del Trasplantament de Medul·la Òssia. Fundació Josep Carreras (contingut en espanyol)
- Què és l’HLA i com funciona? Fundació Josep Carreras (contingut en espanyol)
- La Malaltia Empelt contra Receptor. Fundació Josep Carreras (contingut en espanyol)
- Història del Trasplantament de Medul·la Òssia. Fundació Josep Carreras (contingut en espanyol)
- Com es realitza la cerca d’un donant compatible anònim? Fundació Josep Carreras (contingut en espanyol)
MANUALS DE SUPORT
- Com enfrontar-se a la leucèmia i el limfoma en nens? Leukemia & Lymphoma Society.
- VIURE APRENENT. Protocol d’actuació per a alumnes amb càncer AFANION.
- Guia de suport per a pares de nens oncològics ASION.
- Guia per a joves i adolescents amb càncer ASION.
- Alumnat amb càncer. Guia per a docents ASION.
- La importància del comportament dels pares quan un nen té càncer ASION.
- El meu fill té càncer. Què faig? FARO.
ALIMENTACIÓ
- Com mantenir una alimentació saludable durant el tractament? Fundació Josep Carreras (contingut en espanyol)
- “Buon profit”. Consells dietètics durant el tractament AFANION.
- “Les receptes màgiques de Jabel”. Isabel Rojas Murcia, Carolina Mangas Gallardo.
ALTRES
- Informació sobre els efectes a llarg termini i tardans del tractament per a la leucèmia o el limfoma en els nens Leukemia & Lymphoma Society.
- El meu germà té càncer Fundació Josep Carreras contra la leucèmia. (contingut en espanyol)
- L’escola en un hospital Fundació Josep Carreras contra la leucèmia. (contingut en espanyol)
- Educant il·lusions. Guia per a la intervenció psicoeducativa en nens i adolescents amb càncer FARO.
- El càncer en l’adolescència Fundació Josep Carreras contra la leucèmia. (contingut en espanyol)
- Documental “La leucèmia i els adolescents” Fundació Josep Carreras contra la leucèmia. (contingut en espanyol)
- Documental “Els nadons també tenen leucèmia” Fundació Josep Carreras contra la leucèmia. (contingut en espanyol)
- 7 formes de posar-se un mocador Fundació Josep Carreras contra la leucèmia. (contingut en espanyol)
- Conte “La princesa Luzie i els cavallers de la quimio’ ASPANAFOA.
- Conte “Anem a quimioteràpia’.
- Conte “Anem a radioteràpia’.
- Conte “Gasparín Súper Quimio” Federación Española de Padres de Niños con Cáncer.
- Vídeo “Charlie Brown i la leucèmia”.
- Conte “El Toby i la màquina voladora”.
- Conte “La fada de les estrelles” AECC.
- Conte “La Lina, la petita oreneta” Osakidetza.
Enllaços d'interès: entitats locals (recursos i serveis)
Totes aquestes organitzacions són externes a la Fundació Josep Carreras.
ANDALUSIA
ARAGÓ
ASTÚRIES
CASTELLA-LA MANXA
CASTELLA I LLEÓ
CATALUNYA
COMUNITAT VALENCIANA
EXTREMADURA
GALÍCIA
ILLES BALEARS
ILLES CANÀRIES
LA RIOJA
MADRID
- AAA (asociación de adolescentes y Adultos Jóvenes con Cáncer)
- ASION
- FUNDACIÓN CAICO
- FUNDACIÓN ALADINA
- FUNDACIÓN UNOENTRECIENMIL
MÚRCIA
NAVARRA
PAÍS BASC
Suport i ajuda
Et convidem també a seguir-nos a les nostres xarxes socials principals (Facebook, Twitter i Instagram) on sovint compartim testimonis de superació.Si resideixes a l’Estat espanyol, també pots posar-te en contacte amb nosaltres enviant-nos un correu electrònic a imparables@fcarreras.es perquè t’ajudem a posar-te en contacte amb altres persones que han superat aquesta malaltia.