
¿Qué es la leucemia, la médula ósea y cuáles son los tipos de células sanguíneas?
La leucemia es un tipo de cáncer de las células de la sangre y de la médula ósea. Ver apartado Leucemia, médula ósea y células sanguíneas.
¿Qué es la leucemia mieloide aguda infantil?
 Las leucemias agudas son un grupo de enfermedades neoplásicas caracterizadas por la transformación maligna y producción incontrolada de células hematopoyéticas inmaduras de la línea linfoide (leucemia linfoblástica aguda, LLA) o mieloide (leucemia mieloblástica aguda, LMA).
Las leucemias agudas son un grupo de enfermedades neoplásicas caracterizadas por la transformación maligna y producción incontrolada de células hematopoyéticas inmaduras de la línea linfoide (leucemia linfoblástica aguda, LLA) o mieloide (leucemia mieloblástica aguda, LMA).
En la leucemia mieloide aguda, las células inmaduras de la línea mieloide (mieloblastos) proliferan de forma anormal invadiendo progresivamente la médula ósea e interfiriendo la producción de células normales de la sangre, lo que origina insuficiencia medular e infiltra tejidos extramedulares.
La LMA constituye un 20% de las leucemias diagnosticadas en esta etapa de la vida. La incidencia anual en la edad pediátrica de LMA es de 8 casos por cada millón de niños menores de 15 años (US Cancer Institute ‘s Surveillance, Epidemiology and End Results (SEER) program).
La LMA en el periodo infantil es más frecuente antes de los dos años.
Su incidencia en la etapa escolar desciende y aumenta progresivamente con la edad a partir de la adolescencia.
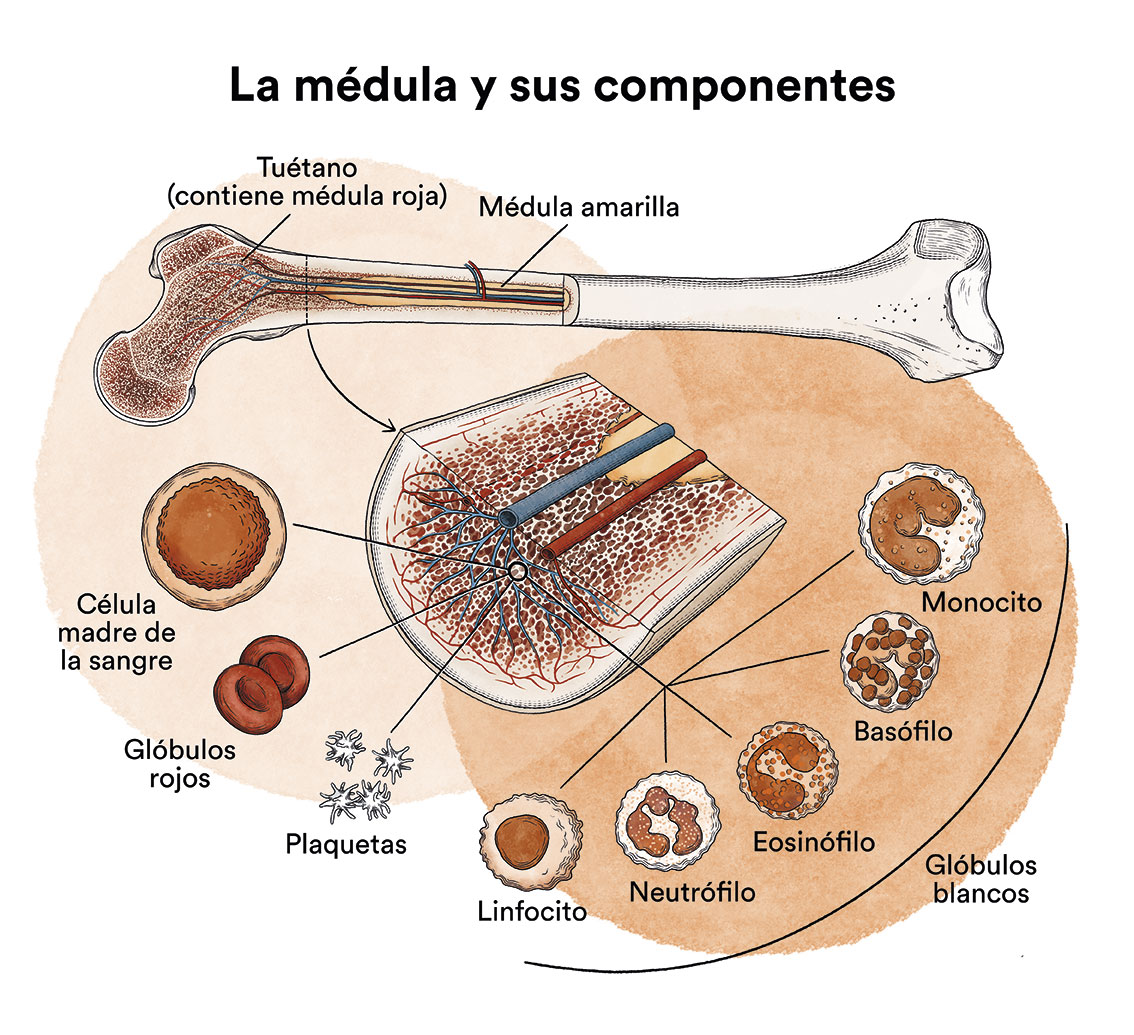
Como hemos visto en ‘Leucemia, médula ósea y células sanguíneas’ , la médula ósea elabora las células madre sanguíneas (células inmaduras) que, con el tiempo, se transformarán en células sanguíneas maduras. Una célula madre sanguínea se convierte en una célula madre mieloide o en una célula madre linfoide.
Una célula madre mieloide se convierte en uno de los tres tipos de glóbulos sanguíneos maduros:
- Glóbulos rojos, que transportan el oxígeno a otros tijos y órganos del cuerpo
- Granulocitos, glóbulos blancos que ayudan a combatir infecciones y otras enfermedades
- Plaquetas, que colaboran en la coagulación de la sangre cuando se produce la rotura de un vaso sanguíneo.
Una célula madre linfoide se convierte en un linfoblasto y, más tarde, en uno de los tres tipos de linfocitos (glóbulos blancos):
- linfocitos B, que producen anticuerpos para ayudar a combatir las infecciones del cuerpo
- linfocitos T, que ayudan a los linfocitos B a producir anticuerpos para combatir infecciones
- linfocitos citotóxicos naturales o linfocitos NK (natural killer), un tipo de célula inmunitaria que contiene enzimas que pueden destruir células tumorales o células infectadas por virus.
En los niños afectados por una leucemia mieloide aguda, hay demasiadas células madre que se transforman en mieloblastos.
¿Cuáles son las causas de la leucemia mieloide aguda infantil?
Las causas específicas que originan la mayoría de los casos de LMA pediátrica no se conocen. Sólo en un porcentaje muy pequeño de casos (alrededor de 5%) las leucemias agudas en la edad pediátrica se desarrollan en pacientes con una enfermedad genética subyacente con predisposición a la leucemia como por ejemplo el síndrome de Down o síndromes congénitas de insuficiencia medular (la Anemia de Fanconi o la Disqueratosis congénita, entre otras).
La leucemia, como otros tipos de cáncer, no es contagiosa. Ver apartado Leucemia, médula ósea y células sanguíneas.
¿Cómo se clasifica la leucemia mieloide aguda infantil?
Los dos esquemas más comúnmente usados para clasificar las LMA son: la clasificación FAB (franco-americana-británica), basada en las características microscópicas y en la expresión de determinadas proteínas en la célula leucémica (inmunofenotipo); y el nuevo sistema de la OMS (Organización Mundial de la Salud), que incorpora información genética o molecular de la célula leucémica y la información clínica con interés pronostico.
En nuestro país, en la práctica clínica habitual, la más extendida es la clasificación FAB.
| FAB | Nombre | % en pediatría |
|---|---|---|
| M0 | LMA con poca diferenciación | 2-5 |
| M1 | LMA sin maduración | 10-15 |
| M2 | LMA con maduración | 25-30 |
| M3 | Leucemia promielocítica aguda | 5-10 |
| M4 | Leucemia mielomonocítica aguda | 15-25 |
| M4Eo | M4 + eosinofilia en la medula ósea | 10 |
| M5 | Leucemia monoblástica/monocítica | 15-25 |
| M6 | Leucemia eritroide | 1-3 |
| M7 | Leucemia aguda megacarioblástca | 5-10 |
La clasificación de la OMS, actualizada en 2022, valora aspectos genéticos y moleculares de las células leucémicas. Las alteraciones citogenéticas más habituales en las LMA son las translocaciones; desplazamiento de un fragmento de un cromosoma a otro cromosoma (se indica como t). Ejemplo: t(8;21), un fragmento del cromosoma 8 se desplaza a una zona del cromosoma 21; o dentro del mismo cromosoma, t(16;16). También pueden observarse inversiones citogenéticas que es cuando un segmento cromosómico cambia de sentido dentro del propio cromosoma (se indica como inv).
 Las translocaciones o inversiones detectadas en los estudios citogenéticos generan reordenamientos de los genes localizados en las regiones cromosómicas afectadas. Éstos se representan por los nombres de los genes implicados, así en el caso de la t(8;21) generará un reordenamiento de los genes RUNX1 y RUNX1T1. Las alteraciones citogenéticas han demostrado ser un factor pronóstico muy importante y son utilizadas en la mayoría de los protocolos de tratamiento para determinar su intensidad.
Las translocaciones o inversiones detectadas en los estudios citogenéticos generan reordenamientos de los genes localizados en las regiones cromosómicas afectadas. Éstos se representan por los nombres de los genes implicados, así en el caso de la t(8;21) generará un reordenamiento de los genes RUNX1 y RUNX1T1. Las alteraciones citogenéticas han demostrado ser un factor pronóstico muy importante y son utilizadas en la mayoría de los protocolos de tratamiento para determinar su intensidad.
En los últimos años se han ido describiendo mutaciones en uno o varios genes de las células leucémicas de la mayoría de los pacientes. Algunas de ellas han demostrado tener importancia pronóstica y ser relevantes para definir la intensidad del tratamiento. Así, en la reciente clasificación de la OMS se incorporan las mutaciones en NPM1 y CEBPA, asociadas a un pronóstico más favorable.
| Clasificación OMS de LMA y neoplasias relacionadas |
|---|
| LMA con alteraciones genéticas recurrentes |
| LMA con t(8;21); RUNX1-RUNX1T1 |
| LMA con inv(16) o t(16;16); CBF-MYH11 |
| LMA con t(15;17); PML-RARA |
| LMA con t(9;11); MLLT3-KMT2A |
| LMA con t(6;9); DEK-NUP214 |
| LMA con inv(3) o t(3;3);GATA2, MECOM |
| LMA (megacarioblástica) con t(1;22); RBM15-MKL1 |
| LMA con mutación en NPM1 |
| LMA con mutación bialélica en CEBPA |
| Entidades provisionales: |
| LMA con BCR-ABL1 |
| LMA con mutación en RUNX1 |
| LMA con cambios en relación a mielodisplasia |
| LMA en relación a tratamiento |
| LMA no específica (NOS) |
| LMA con poca diferenciación |
| LMA sin maduración |
| LMA con maduración |
| Leucemia mielomonocítica aguda |
| Leucemia monoblástica/monocítica |
| Leucemia eritroide |
| Leucemia aguda megacarioblástica |
| Leucemia aguda basofílica |
| Panmielosis con mielofibrosis aguda |
| Sarcoma mieloide |
| Proliferación mieloide en relación a Síndrome de Down |
| Mielopoyesis anómala transitoria |
| Leucemia mieloide asociada a Síndrome de Down |
| Leucemias agudas de linaje ambiguo |
La mayoría de los casos en pediatría se clasifican dentro del grupo de LMA con alteraciones genéticas recurrentes o LMA no específica.
¿Cuáles son los síntomas de la leucemia mieloide aguda infantil?
La presentación clínica de la leucemia mieloide aguda es variable y, en general, los síntomas en el diagnóstico se deben a la infiltración de las células leucémicas de la médula ósea y otros órganos. Aunque puede presentarse de forma insidiosa, la LMA infantil suele hacerlo de forma aguda, con una historia de menos de tres meses desde el inicio de la clínica hasta el diagnóstico.
En la leucemia mieloide aguda, la producción de las células sanguíneas normales se ve alterada por el crecimiento de las células leucémicas en la médula ósea. Esto puede ocasionar:
- Cansancio, debilidad, mareos y palidez (por anemia)
- Aparición de morados y pequeñas manchas rosadas en la piel (petequias) u otros sangrados (por un recuento de plaquetas bajo): hemorragias nasales, de encías o de cualquier otro foco.
- Fiebre e infecciones que no evolucionan bien (debido al mal funcionamiento de los leucocitos)
En algunas ocasiones ocurre el crecimiento de los ganglios linfáticos, el hígado o el bazo. Puede, asimismo, observarse sintomatología especifica de la infiltración del sistema nervioso central (dolor de cabeza, vómitos, somnolencia, etc.), piel (nódulos diseminados o zonas de piel engrosada), mucosas (inflamación de las encías), ocular (visión borrosa, ceguera), entre otras.

Al inicio de la enfermedad, todos estos síntomas pueden ser muy parecidos a los de una infección por un virus. Cuando los síntomas continúan más de 2-4 semanas, en una mayoría de casos se puede llegar a hacer el diagnóstico. Como no son síntomas específicos o exclusivos de la leucemia, es muy frecuente que se haya consultado en diversas ocasiones al médico antes de que se llegue al diagnóstico. Generalmente, esto no influye en las opciones de curación del niño o niña.
¿Cómo se diagnostica la leucemia mieloide aguda infantil?
Además de los estudios básicos en sangre y medula ósea (morfología, recuento, inmunofenotipo) a realizar en toda leucemia, los estudios citogenéticos (para detectar anomalías cromosómicas concretas) y estudios moleculares (para detectar alteraciones genéticas especificas) son fundamentales para tipificar y clasificar la enfermedad. Determinadas alteraciones genéticas y moleculares se correlacionan con la sensibilidad al tratamiento y al riesgo de recaída.
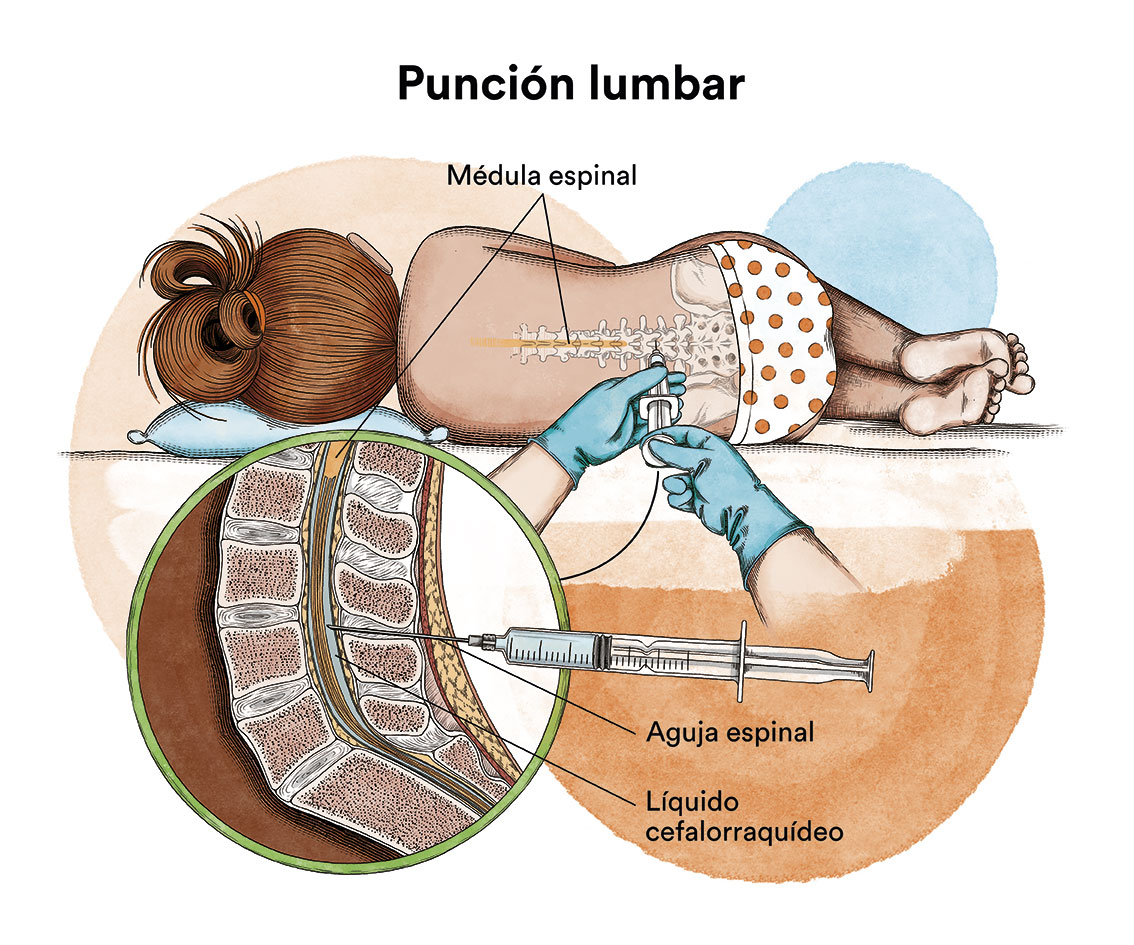
También debe estudiarse si la enfermedad se ha extendido al sistema nervioso central efectuando, para ello, una punción lumbar con el fin de analizar el líquido que envuelve dicho sistema (líquido cefalorraquídeo).
¿Cuál es el tratamiento de la leucemia mieloide aguda en niños?
 El objetivo del tratamiento de la leucemia mieloide aguda es eliminar las células leucémicas para permitir que la médula ósea vuelva a trabajar con normalidad.
El objetivo del tratamiento de la leucemia mieloide aguda es eliminar las células leucémicas para permitir que la médula ósea vuelva a trabajar con normalidad.
El pronóstico de las LMA en pediatría ha mejorado de forma significativa en las últimas décadas. Esta mejora viene dada, entre otros, por una mejor clasificación o estratificación de cada paciente en grupos de riesgo, es decir, según el riesgo individual de recaída. Esta estratificación permite aplicar estrategias terapéuticas adaptadas, por lo que se intensificará el tratamiento en los pacientes que tienen unos factores pronósticos de Alto Riesgo y se reducirá en aquellos de Bajo Riesgo de recaída.
El objetivo final del tratamiento es conseguir la remisión completa de la enfermedad y que ésta sea profunda (a nivel molecular) y permanente.
Diferenciamos básicamente dos fases de tratamiento: de inducción y de post-remisión o consolidación. La fase de mantenimiento con dosis bajas de quimioterapia utilizada en los protocolos de leucemia linfoblástica aguda ha sido abandonada en la mayoría de los protocolos de LMA por no aportar una eficacia adicional, exceptuando en el subgrupo de la leucemia promielocítica aguda (ver a continuación).
El tratamiento de la leucemia mieloide aguda pediátrica se basa siempre en quimioterapia intensiva; administración endovenosa de distintos fármacos citostáticos (quimioterapia) a lo largo de varios ciclos de tratamiento. Habitualmente, aunque puede variar según el protocolo, se realizan 1 o 2 ciclos de inducción seguidos de 2-3 ciclos de consolidación.

El tratamiento de inducción persigue la eliminación de las células leucémicas de la sangre y de la mayor parte de enfermedad presente en la medula ósea y así restituir su funcionamiento normal, a este hecho se le denomina alcanzar la remisión completa. Esta situación clínica suele alcanzarse tras el primer ciclo de inducción, si bien en ocasiones puede ser necesario administrar dos ciclos de inducción para alcanzar el estado de remisión completa. Con los protocolos actuales, más de un 85% de los pacientes alcanzaran la remisión completa tras la fase de inducción.
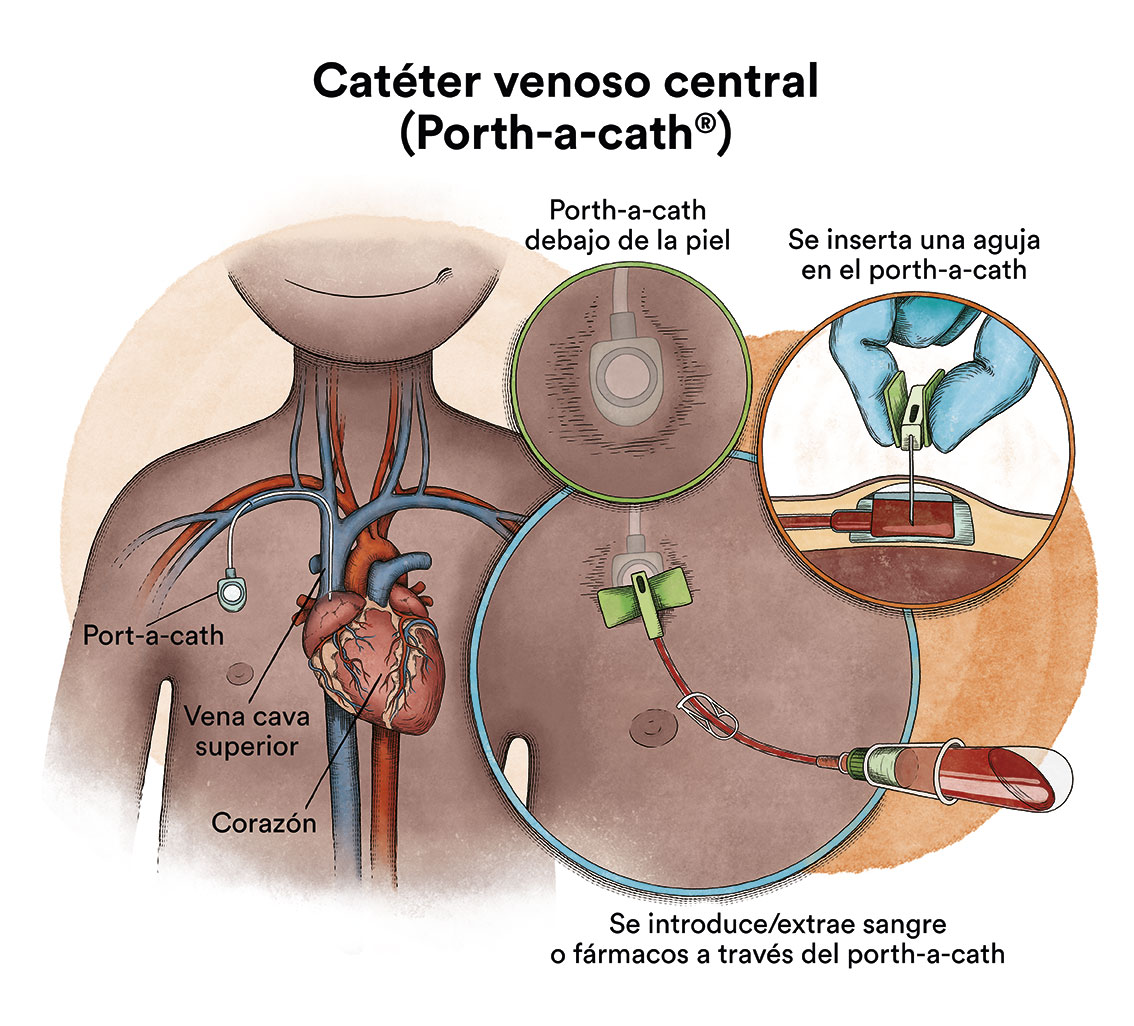
Cuando la quimioterapia se administra por vía intravenosa, para evitar pinchar repetidamente una vena, se utiliza un dispositivo especial llamado catéter. El catéter se introduce en una vena grande que permite administrar todo tipo de medicamentos, así como también extraer sangre para los análisis de sangre, evitando las repetidas punciones al pequeño.
Existe un tipo de catéter, llamado port-a-cath, que se une a un reservorio redondo de plástico o metal que queda bajo la piel del tórax. El port-a-cath es muy práctico en niños porque al quedar bajo la piel no permite que el niño o niña se lo arranque, es más difícil que se infecte que otros tipos de catéter y permite que el niño se bañe.
A continuación, debe efectuarse un tratamiento de post-remisión o consolidación que tiene por objetivo eliminar las células leucémicas residuales (enfermedad residual mínima), potenciales responsables de la recaída de la enfermedad.
Si la quimioterapia se administra por un catéter venoso llega, por la sangre, a la casi totalidad de las células del cuerpo. Sin embargo, la mayoría de los medicamentos de la quimioterapia no llegan bien al líquido cefalorraquídeo que baña el cerebro y médula espinal. Esto hace que haya células leucémicas que pueden sobrevivir en este líquido. Con el fin de prevenir que las células leucémicas que llegan al líquido cefalorraquídeo sobrevivan y sean la causa de una futura recaída al sistema nervioso, se debe administrar quimioterapia directamente en el líquido cefalorraquídeo, mediante punciones lumbares (quimioterapia intratecal). La utilización de radioterapia craneal ha sido abandonada en la mayoría de los protocolos.
En algunos pacientes puede estar indicado realizar un trasplante de progenitores hematopoyéticos (TPH) (comúnmente conocido como trasplante de médula ósea) como parte del tratamiento de consolidación. La mayoría de los protocolos actuales han abandonado el autotrasplante de medula ósea (de uno mismo) para esta patología y, en caso de estar indicado, se recomienda el TPH procedente de un donante compatible (TPH alogénico), ya sea familiar o no relacionado.
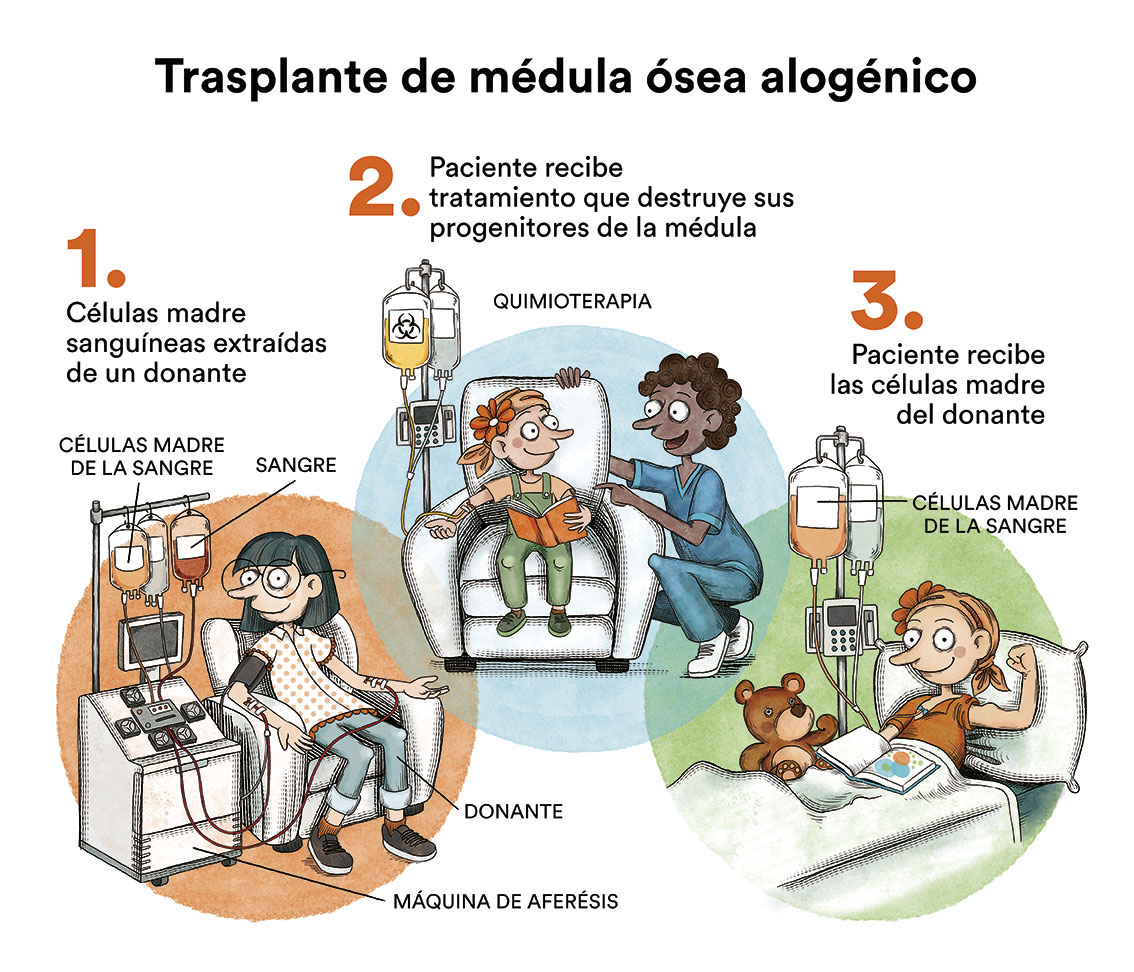
El TPH en LMA pediátrica es un tema en constante revisión; las indicaciones de TPH en pacientes con LMA en primera remisión completa (pacientes que no han presentado una recaída) son controvertidas y no son las mismas en todos los países. En general, este tratamiento queda reservado para aquellos casos considerados de alto riesgo por las características biológicas de la enfermedad o por una inadecuada respuesta al tratamiento quimioterápico. La mayoría de los pacientes que han recaído y logran un segundo estado de remisión completa son candidatos a TPH.

¿Qué probabilidades tienen de curarse los niños con leucemia mieloide aguda?
Las probabilidades de curación vienen determinadas por las características del paciente, de la enfermedad (alteraciones genéticas/moleculares), el tratamiento que se administre y por la respuesta que se presente a éste. A diferencia de los adultos, dónde las características del paciente, como la edad avanzada o la co-existencia de otras patologías, suelen ser muy relevantes, éstos no son factores relevantes en la edad pediátrica.
La supervivencia de los niños con leucemia mieloide aguda ha mejorado notablemente en los últimos años, con tasas de supervivencia de alrededor del 65%. Esta mejoría ha sido posible gracias al incremento en la intensidad del tratamiento quimioterápico, una mejor clasificación de los pacientes en grupos de riesgo, la implementación de medidas de soporte más eficaces (mejores antibióticos, facilidad para las transfusiones de sangre y plaquetas, soporte nutricional, enfermería especializada ..), así como una notable mejora en la selección de donantes para la realización del TPH.
Nuevos tratamientos contra la leucemia mieloide aguda infantil
En los últimos años se está avanzando hacia tratamientos más personalizados donde se tengan en cuenta las características de cada individuo y de la enfermedad (subtipo genético, molecular, …). La investigación en este campo es muy activa y, por tanto, no es de extrañar que se hayan desarrollado nuevos fármacos para estas enfermedades. La mayoría de ellos aún no forman parte de los protocolos estándar de tratamiento, pero muchos se encuentran en fases avanzadas de implementación clínica.
Dentro de las distintas líneas de desarrollo de nuevos fármacos, cabe destacar:
- Nuevos fármacos quimioterápicos: Actúan de manera similar a los fármacos existentes, pero tienen una mayor eficacia y/o menor toxicidad. Por ejemplo, la daunorrubicina liposomal permite administrar dosis altas de tratamiento y, por tanto, muy eficaces, pero con una baja toxicidad sobre el corazón, uno de los principales inconvenientes de este grupo de fármacos.
- Terapias dirigidas: Son fármacos que están dirigidos hacia componentes específicos de las células tumorales y tienen un menor impacto sobre las células sanas. Dentro de este grupo destacamos:
- Los anticuerpos monoclonales, combinan un fármaco antineoplásico con un anticuerpo que reconoce proteínas de la célula tumoral. La identificación de alteraciones citogenéticas-moleculares en la mayoría de los pacientes con LMA ha permitido el desarrollo de nuevos fármacos que, por distintos mecanismos, actúan sobre estas dianas «moleculares» específicas y, por tanto, son muy selectivos sobre las células neoplásicas.
- La inmunoterapia aprovecha las propiedades del sistema inmunológico propio para actuar sobre las células leucémicas. Es una de las áreas de mayor investigación en los últimos años, pero aun con escasa aplicación en LMA.
Subtipos específicos
- Leucemia Promielocítica Aguda
Una de las leucemias que más se ha beneficiado de una estrategia terapéutica individualizada es la leucemia aguda promielocítica. En las últimas décadas, gracias a la investigación científica, se ha obtenido una mejora sustancial en su tratamiento, pasando de ser un subtipo de LMA con muy mal pronóstico a ser una enfermedad que responde muy bien al tratamiento. Este tipo de leucemia se caracteriza porque tiene una translocación entre los cromosomas 15 y 17 [t(15:17)], que afecta al receptor del ácido retinoico alfa (RARα o RARA) y que le confiere una alta sensibilidad al tratamiento con ácido holotransretinoico (ATRA).
- Pacientes con Síndrome de Down
Los niños con síndrome de Down tienen un riesgo 15 veces superior a presentar una leucemia aguda. En el caso de las LMA, la edad de presentación suele ser por debajo de los 5 años y de forma característica el subtipo que presentan son leucemia megacarioblástica aguda (M7, según clasificación FAB) o leucemia eritroide aguda (M6, según clasificación FAB).
Este grupo de pacientes presenta una elevada sensibilidad a los tratamientos quimioterápicos y esto ha posibilitado tasas de curación elevadas. Una de las principales dificultades para lograr la curación se debe a la elevada toxicidad ante algunos fármacos quimioterápicos y el elevado riesgo de infección. Es por ello por lo que diferentes grupos han conseguido aumentar la supervivencia con protocolos de tratamientos adaptados.
Hasta un 10% de los niños con síndrome de Down presentan una proliferación transitoria de células leucémicas durante los primeros meses de vida. Estas células son morfológicamente indistinguibles de una LMA. Este fenómeno se conoce como síndrome mieloproliferativo transitorio o mielopoyesis anómala transitoria. Habitualmente presentan un curso benigno y suelen involucionar espontáneamente durante los tres primeros meses de vida, aunque algunos pacientes pueden requerir tratamientos con dosis bajas de citostáticos. Es importante el seguimiento posterior, ya que un 20% de estos niños desarrollarán una LMA durante los tres primeros años de vida.
Seguimiento
Después de completar el tratamiento, el niño seguirá controles periódicos por su médico hematólogo y por otros especialistas en caso necesario. Los controles se realizan para evaluar una posible recaída y para hacer un seguimiento y un tratamiento de las posibles complicaciones a largo plazo. Estos controles se van espaciando progresivamente hasta hacerse una vez al año. Es recomendable realizar un seguimiento como mínimo anual a largo plazo para poder detectar pronto y poder tratar, si aparecieran, las secuelas del tratamiento o de la leucemia.
Recomendaciones y otros aspectos prácticos
A continuación, hacemos unas recomendaciones de carácter general y que responden a algunas de las preguntas más frecuentes que realizan los padres de los niños con leucemia:
- ¿Se le caerá el pelo? ¿Cuándo? ¿Lo debemos cortar?
Con la quimioterapia que recibirá para tratar la leucemia, el cabello se le caerá. Generalmente, esto sucede a las 2-3 semanas del inicio de la quimioterapia. Si el niño o la niña tiene el pelo largo, es más adecuado cortarlo corto antes de que empiece a caer. No es necesario, ni conveniente desde el punto de vista psicológico, cortarlo durante los primeros días del ingreso. Tampoco hay que explicarle este hecho en un primer momento. Sí conviene, sin embargo, abordar este tema con el niño antes de que empiece a caer. El cabello vuelve a salir al cabo de 2-4 semanas de haber iniciado la fase de tratamiento de mantenimiento, en la que la quimioterapia es de menor intensidad.
- Higiene
Debido a que el niño tiene disminuidas sus defensas ante las infecciones (por la propia enfermedad y también por el tratamiento administrado), es conveniente mantener una higiene corporal adecuada del niño, de la habitación del hospital y del domicilio familiar, así como de sus juguetes.
Es recomendable evitar aquellos juguetes que almacenen mucho polvo y las cajas de cartón. Tampoco se ha de almacenar comida fuera del frigorífico. Las plantas están prohibidas en la habitación, ya que en la tierra hay esporas de hongos.
El orden facilita la limpieza por parte del personal de la limpieza del hospital.
- Visitas
Es conveniente reducir el número de visitas en la habitación del niño, ya que pueden ser portadoras de infecciones. Es recomendable que no haya más de 2 acompañantes en la habitación y que se laven las manos antes de entrar. Si alguno de los visitantes tiene algún proceso infeccioso (resfriado, conjuntivitis …) es preferible que no venga.
En el caso de que éste sea el padre, la madre u otra persona que cuide del niño y que no se pueda prescindir de su atención, convendría que se pusieran una mascarilla y se lavaran las manos antes de entrar en contacto con el niño.
- Alimentación
El niño que recibe tratamiento con quimioterapia intensiva debe recibir una alimentación variada. Es conveniente, cuando la cifra de leucocitos es baja, evitar los alimentos crudos que no se puedan pelar (por ejemplo: lechuga, fresas, tomate crudo).
En ocasiones la quimioterapia puede quitar el hambre, o incluso provocar náuseas. Durante los días que esté recibiendo la quimioterapia, no conviene forzar al niño para que coma, porque puede ser contraproducente.
Por otro lado, los corticoides (prednisona y dexametasona) que recibirá en algunas fases del tratamiento pueden aumentar mucho el apetito, incluso con ansiedad. Si bien se les puede permitir comer algo más que las comidas que se sirven en el hospital, no se les debe dejar comer sin límite, ya que con frecuencia no lo toleran bien y puede ocasionar dolor de estómago.
Enlaces de interés sobre temas médicos relacionados con la leucemia mieloide aguda en niños
- Tratamiento de la leucemia mieloide aguda en niños. National Cancer Institute.
- Información sobre la leucemia promielocítica aguda. Leukemia and Lymphoma Society
- La leucemia mieloide aguda en niños y adolescentes. Leukemia and Lymphoma Society
Enlaces de interés sobre otros temas relacionados con la leucemia mieloide aguda en niños
MATERIALES LEUCEMIA INFANTIL
- Los bebés también tienen leucemia. Fundación Josep Carreras contra la leucemia.
- Leucemia infantil. Los pequeños imparables. Fundación Josep Carrerascontra la leucemia.
- Juego recortable Medulín. Fundación Josep Carreras contra la leucemia.
La Fundación Josep Carreras dispone de un cuento “El bebé forzudo” dirigido a niños o hermanos que padecen leucemia. Está especialmente dirigido a niños hasta los 6 años. Si quieres solicitarlo, puedes enviarnos un correo a imparables@fcarreras.es.
TRASPLANTE DE MÉDULA ÓSEA
- Guía del Trasplante de Médula Ósea. Fundación Josep Carreras contra la leucemia.
- ¿Qué es el HLA y cómo funciona? Fundación Josep Carreras contra la leucemia.
- La Enfermedad Injerto contra Receptor Fundación Josep Carreras contra la leucemia.
- Historia del Trasplante de Médula Ósea. Fundación Josep Carreras contra la leucemia.
- ¿Cómo se realiza la búsqueda de un donante compatible anónimo? Fundación Josep Carreras contra la leucemia.
- Guía de cuidados para niños trasplantados. TransplantCHild.
- El trasplante de células madre: un libro para colorear. Leukeamia and Lymphoma Society.
MANUALES DE APOYO
- ¿Cómo enfrentarse a la leucemia y el linfoma en niños? Leukemia & Lymphoma Society.
- VIVIR APRENDIENDO. Protocolo de actuación para alumnos con cáncer AFANION.
- Guía de apoyo para padres de niños oncológicos ASION.
- Guía para jóvenes y adolescentes con cáncer ASION.
- Alumnado con cáncer. guía para docentes ASION.
- La importancia del comportamiento de los padres cuando un niño tiene cáncer ASION.
- Mi hijo tiene cáncer. ¿Qué hago? FARO.
ALIMENTACIÓN
- ¿Cómo mantener una alimentación saludable durante el tratamiento? Fundación Josep Carreras contra la leucemia.
- “Buen provecho”. Consejos dietéticos durante el tratamiento AFANION.
- ‘Las recetas mágicas de Jabel’. Isabel Rojas Murcia, Carolina Mangas Gallardo.
OTROS
- Información sobre los efectos a largo plazo y tardíos del tratamiento para la leucemia o el linfoma en los niños Leukemia & Lymphoma Society.
- Mi hermano tiene cáncer Fundación Josep Carreras contra la leucemia.
- La escuela en un hospital Fundación Josep Carreras contra la leucemia.
- Educando ilusiones. Guía para la intervención psicoeducativa en niños y adolescentes con cáncer FARO.
- El cáncer en la adolescencia Fundación Josep Carreras contra la leucemia.
- Documental ‘La leucemia y los adolescentes’ Fundación Josep Carreras contra la leucemia.
- Documental ‘Los bebés también tienen leucemia’ Fundación Josep Carreras contra la leucemia.
- 7 formas de ponerse un pañuelo Fundación Josep Carreras contra la leucemia.
- Cuento ‘La princesa Luzie y los caballeros de la quimio’ ASPANAFOA.
- Cuento ‘Vamos a quimioterapia’.
- Cuento ‘Vamos a radioterapia’.
- Cuento ‘Gasparín Super Quimio’ Federación Española de Padres de Niños con Cáncer.
- Vídeo ‘Charlie Brown y la leucemia’.
- Cuento ‘Toby y la máquina voladora’.
- Cuento ‘El hada de las estrellas’ AECC.
- Cuento ‘Lina la pequeña golondrina’ Osakidetza.
Enlaces de interés: entidades locales (recursos y servicios)
Todas estas organizaciones son externas a la Fundación Josep Carreras.
ANDALUCÍA
ARAGÓN
ASTURIAS
CASTILLA LA MANCHA
CASTILLA LEÓN
CATALUÑA
COMUNIDAD VALENCIANA
EXTREMADURA
GALICIA
ISLAS BALEARES
ISLAS CANARIAS
LA RIOJA
MADRID
- AAA (asociación de adolescentes y Adultos Jóvenes con Cáncer)
- ASION
- FUNDACIÓN CAICO
- FUNDACIÓN ALADINA
- FUNDACIÓN UNOENTRECIENMIL
MURCIA
NAVARRA
PAÍS VASCO
Apoyo y ayuda
Te invitamos también a seguirnos a través de nuestras redes sociales principales (Facebook, Twitter e Instagram) en las que, a menudo, compartimos testimonios de superación.
Si resides en España, también puedes ponerte en contacto con nosotros enviándonos un correo electrónico a imparables@fcarreras.es para que te ayudemos a ponerte en contacto con otras familias que han superado esta enfermedad.