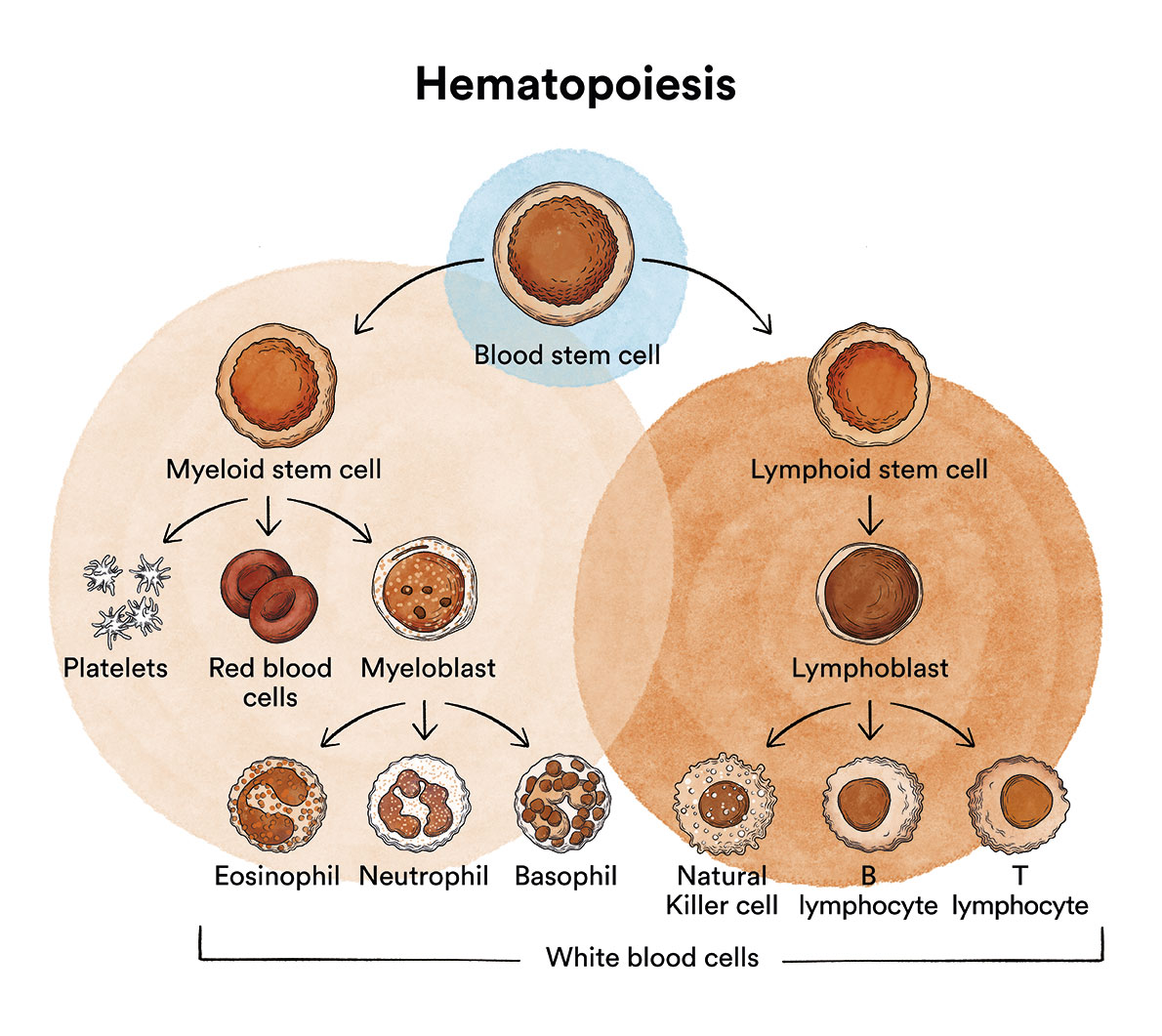
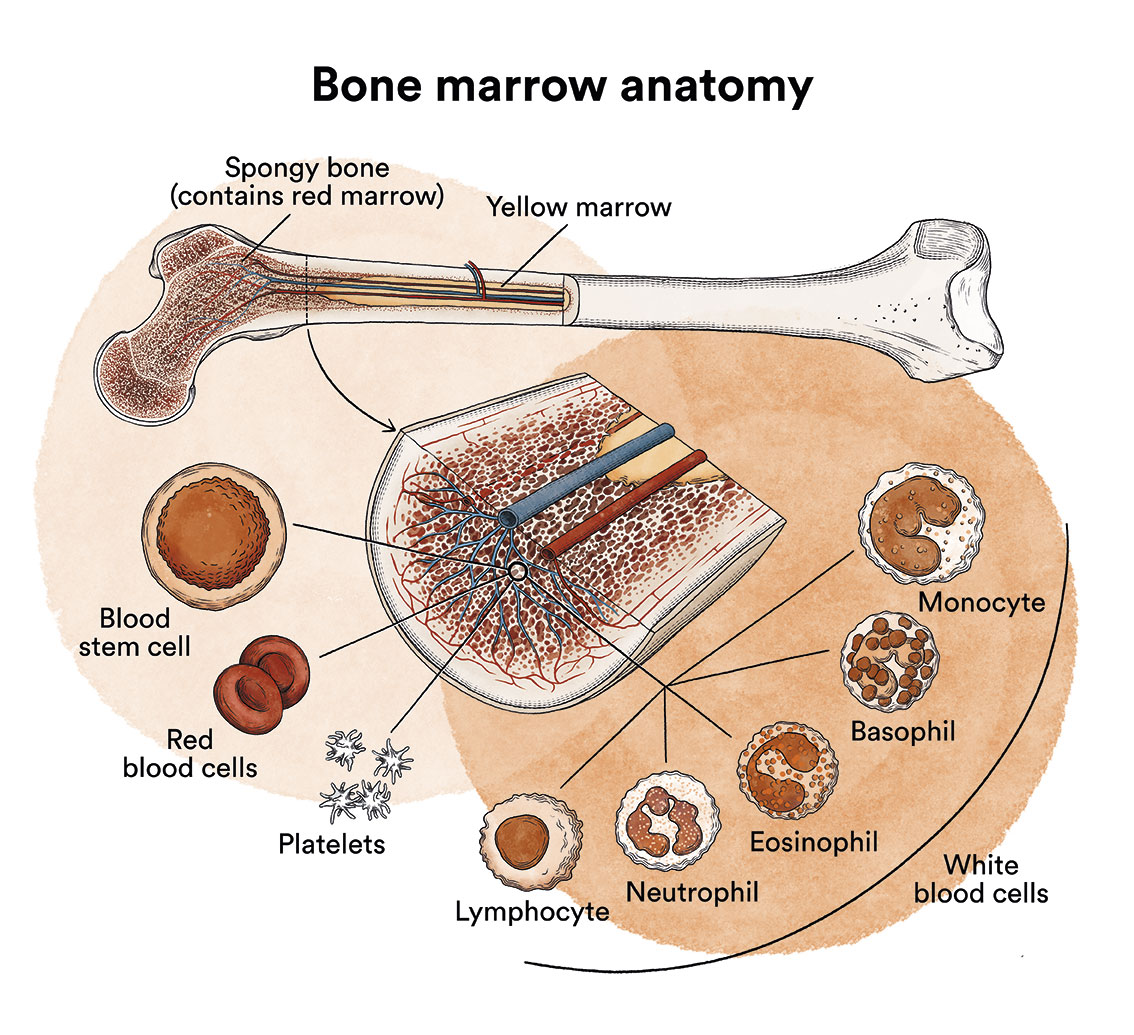
How does bone marrow work and what are the types of blood cells?
Aplastic Anemia is a disease that originates in the bone marrow but, unlike leukaemia or lymphoma, it is not a cancer. See Leukaemia, bone marrow and blood cells.
What is aplastic anemia and who is affected?
Aplastic anemia is a disease in which the red bone marrow disappears and consequently ceases to produce red blood cells, white blood cells and platelets.
Aplastic anemias are classified into two groups: congenital or, more commonly, acquired. Aplastic anemia caused by a congenital alteration (Fanconi anaemia, dyskeratosis congenita), is less frequent than acquired aplastic anemia and is caused by a genetic change that can affect other organs. Patients therefore frequently present other alterations which may be visible from birth.
Aplastic anemia is considered a rare disease, i.e. the percentage of the population that is diagnosed with it and suffers from it is very low (< 0.05%). In Europe and North America the estimated incidence is between 1.5 and 2 new cases per million, per year. However this incidence may be up to three times greater in certain geographical areas such as the Far East, southwest Asia (Japan and China) and Mexico.
Acquired aplastic anemia generally affects teenagers and young adults (15-25 years of age) and people over the age of 60. It affects both male and female equally.
What are the causes of aplastic anemia?
It is not known why aplastic anemia occurs. Acquired aplastic anemia is caused by the disappearance of haematopoietic stem cells, either secondary to direct damage caused by a toxic substance or by an immunological mechanism in the patient’s own lymphocytes, which recognise their stem cells as foreign and destroy them. Although acquired aplastic anemia can sometimes be related to a trigger, the ingestion of a drug, contact with chemical agents, pesticides, benzene, paints or ionising radiation or having suffered certain viral infections (hepatitis being the most frequent), in most cases (80%) it is not possible to identify the cause.
Aplastic anemia is not contagious.
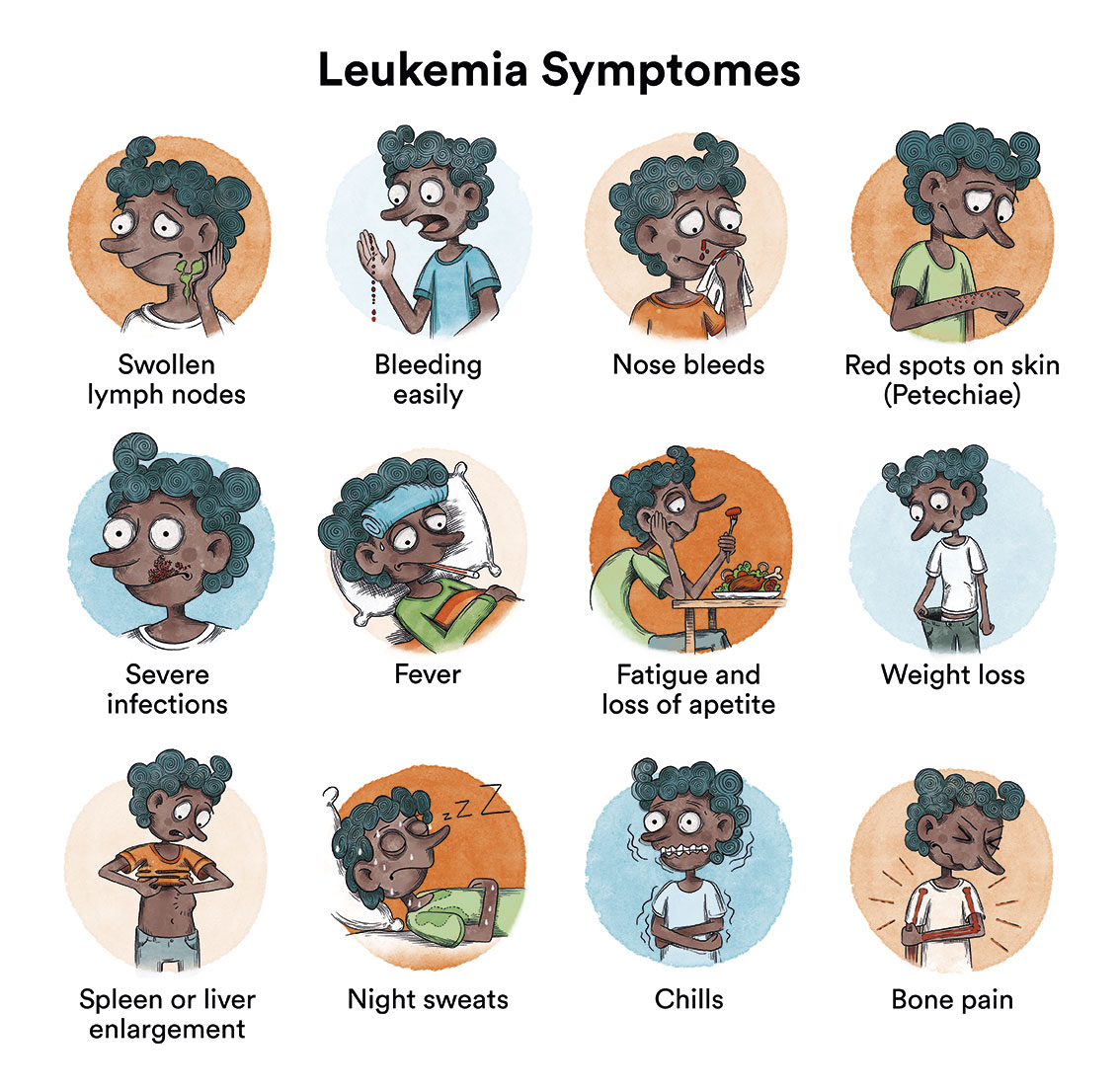
What are the symptoms of aplastic anemia?
The bone marrow works like a factory. It produces red blood corpuscles (or red blood cells, responsible for transporting oxygen to the tissues), leukocytes (or white blood cells, responsible for defending the body against infection), and platelets (responsible for avoiding haemorrhages).
With aplastic anemia, as a consequence of the failure to produce these elements of the blood, patients may present various symptoms, and to different degrees, depending on the deficit. A deficit of red blood cells (anaemia) can manifest itself as tiredness, weakness, paleness, dizziness, palpitations and headache, although, in general, anaemia is usually well tolerated because it establishes itself slowly and progressively. A deficit of white blood cells (leukopenia and neutropenia) can lead to mouth ulcers and continuous infections. A deficit of platelets (thrombopenia) can lead to bruising after the slightest knock, bleeding of the gums, nose or conjunctiva, as well as more serious bleeding from any other parts of the body.
How is aplastic anemia diagnosed?
A blood test (haemogram) to study some of the symptoms mentioned previously, can lead to a suspicion of aplastic anemia. Usually the blood count shows low numbers of red blood cells, white blood cells and platelets (pancytopenia), however, diagnosis can not be confirmed until there has been a study of the bone marrow architecture by means of a biopsy. A bone marrow biopsy is a straight forward procedure in which, by means of puncturing the hip, a small cylinder of bone is obtained. At the same time the biopsy is performed a blood sample is also taken from within the bone marrow (aspiration) and this is used to complement the biopsy studies in order to differentiate between acquired aplastic anemia and other causes of pancytopenia.
It is possible to talk about less severe, severe and very severe aplastic anemia, depending on how intensely the bone marrow is affected.
What is the treatment for aplastic anemia?
- Support treatment: The aim of support treatment is to alleviate the patient’s symptoms. In order to improve the manifestations deriving from the anaemia, patients are administered a red blood cell transfusion. If there is secondary bleeding due to the lack of platelets, a platelet transfusion is administered. In cases where there is fever, a course of antibiotics is administered and, given that such patients have low defences, the necessary levels of hygiene must be observed to avoid further infections. Patients with less severe aplasia may almost be asymptomatic and not require such support treatment.
Transfusion support should be restricted in these patients, especially if they are candidates for allogeneic transplantation, as transfusions predispose to the development of antibodies in the patient that may hinder the implantation of new bone marrow.
- Targeted therapy: In cases where the cause of aplastic anemia has been identified, the main treatment is to suppress the causal agent. Sometimes androgens (male hormones) are used to correct the aplasia, however, it must be borne in mind that these drugs might have significant side effects and are more effective in those aplasias associated with protein defects that keep the size of the chromosomes stable (telomerase). As mentioned at the beginning, acquired aplastic anemia presents secondary to an autoimmune mechanism and patients are treated with immunosuppressive drugs in association, on occasion, with a stem cell transplant.
Patients with less severe aplasia must be treated with an immunosuppressive agent called cyclosporin A in association, sometimes, with anti-thymocyte globulin (better known as ATG) and factors to stimulate haematopoiesis (such as G-CSF). Symptoms are resolved with this treatment for 50-60% of patients although, in a third of cases the aplasia may reappear after time, it being necessary to repeat the treatment.
The treatment of choice, for severe and very severe aplastic anemias for patients under the age of 50, is a bone marrow transplant from a sibling or histocompatible (HLA identical) non-related donor. If an identical family donor cannot be found, bone marrow transplant from an unrelated donor is still the treatment of choice until the age of 20, and would be the second choice between the ages of 20 and 50, in case of non-response to immunosuppressive treatment.
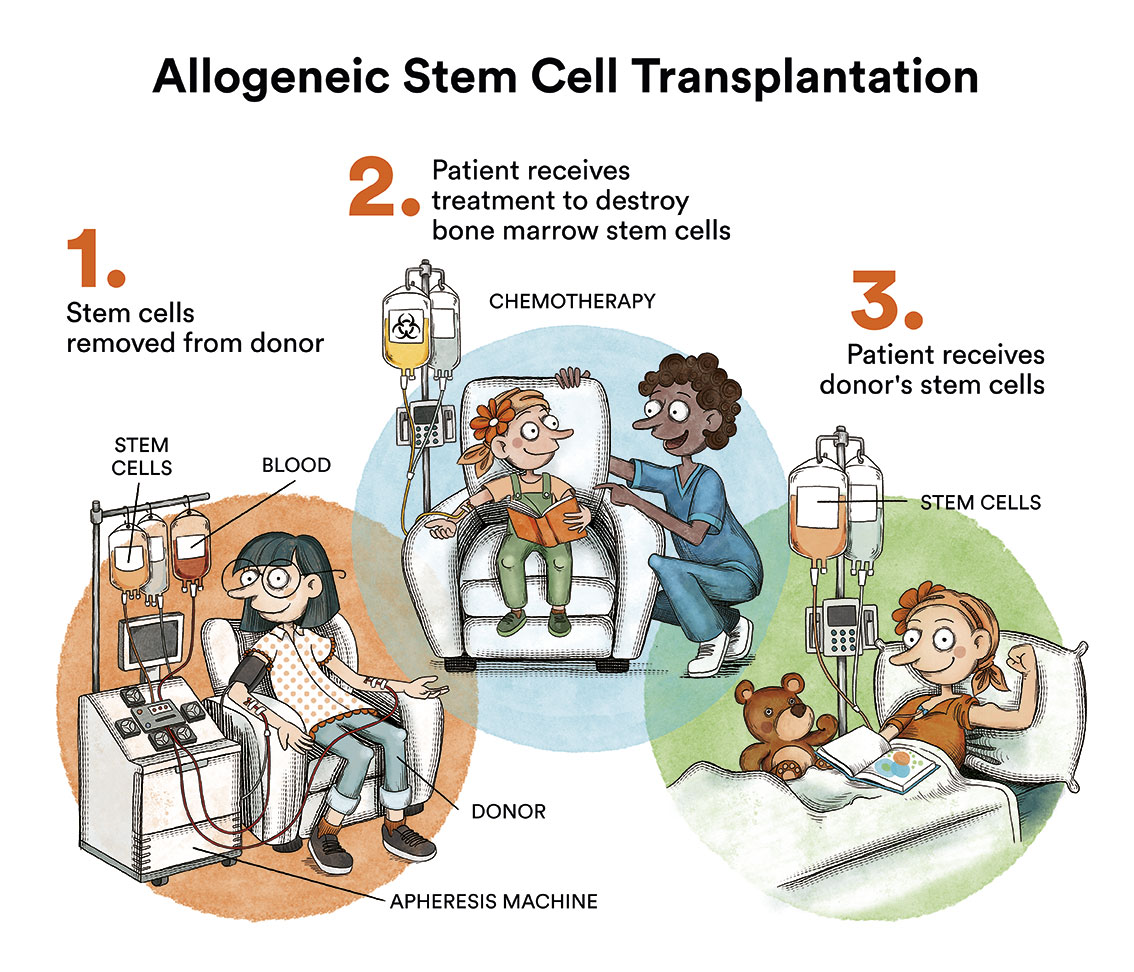
In patients over 50 years of age, first-line immunosuppressive treatment is generally recommended, although, if an HLA-identical family donor is available, it would be possible to consider transplantation as first-line, on an individual basis, depending on the patient’s characteristics. The immunosuppressive treatment offered to patients who do not receive a transplant as first line, because they are older or do not have an HLA compatible donor, is based on the administration of anti-thymocyte globulin (ATG) associated with cyclosporine A. ATG should always be administered on an in-patient basis, as the patient will need central venous access to receive it and infusion reactions (fever, chills, hypotension) are frequent, so the infusion is often prolonged for at least 6 hours. These drugs resolve aplasia in 50-70% of cases, although a third of patients will relapse some time later and the responses obtained are mostly partial (haemogram does not normalise). Although it is still pending authorisation by health agencies, a recent study has observed that the addition of a haematopoiesis-stimulating drug (eltrombopag) at the start of treatment with ATG and cyclosporin A increases the number of overall and complete responses. The worst responses occur mainly in patients older than 40 years and with very severe aplastic anemia.
If the patient relapses or does not respond to initial treatment (patients who have not responded 4-6 months after receiving immunosuppressive treatment), a second cycle of immunosuppressive treatment should be considered (30% response rate), the administration of eltrombopag (45-50% response rate) or a bone marrow transplant if there is a compatible donor (family or unrelated) or even with a haploidentical donor (who shares half of the patient’s identity).
What is the prognosis for patients with aplastic anemia?
The long-term prognosis depends on the severity of the aplasia, the patient’s age, earliness in starting the immunosuppressive treatment and the availability, or otherwise, of a compatible (related or non-related) donor. Two thirds of patients who receive immunosuppressive treatment are cured after one or two courses. With a bone marrow transplant the probability of a cure varies between 70-95% for transplants from compatible siblings, and 70-85% for transplants from non-related donors.
Links of interest concerning medical issues relating to aplastic anemia
Aplastic Anemia. Cleveland Clinic
Links of interest on other topics related to aplastic anemia
TESTIMONIAL MATERIALS
You can order the booklets in paper format for free delivery in Spain by e-mail: imparables@fcarreras.es
BONE MARROW TRANSPLANT
- Bone Marrow Transplant Guide. Josep Carreras Foundation (content in Spanish)
- What is HLA and how does it work? Josep Carreras Foundation (content in Spanish)
- Graft-versus-Host Disease. Josep Carreras Foundation (content in Spanish)
- History of Bone Marrow Transplantation. Josep Carreras Foundation (content in Spanish)
- How is the search for an anonymous donor conducted? Josep Carreras Foundation (content in Spanish)
FOOD
- How to maintain a healthy diet during treatment? Josep Carreras Foundation (content in Spanish)
- Nutrition guide. Leukemia & Lymphoma Society
OTHER
- Ideas on what to take with me to the isolation chamber. Josep Carreras Leukaemia Foundation (content in Spanish)
- Travel tips for people with cancer. Josep Carreras Leukaemia Foundation (content in Spanish)
- Physiotherapy manual for haematological and transplant patients. Josep Carreras Leukaemia Foundation (content in Spanish)
- Prevention and treatment of oral mucositis. Josep Carreras Leukaemia Foundation (content in Spanish)
- Oral hygiene in oncohaematological patients. Josep Carreras Leukaemia Foundation (content in Spanish)
- Fertility manual: Suffering from blood cancer and becoming a parent. Josep Carreras Leukaemia Foundation (content in Spanish)
- Skin care in the oncohaematological patient. Josep Carreras Leukaemia Foundation (content in Spanish)
- Aesthetic Oncology Manual. Josep Carreras Leukaemia Foundation (content in Spanish)
- Leukaemia and sexuality. Josep Carreras Leukaemia Foundation (content in Spanish)
- 7 ways to wear a scarf. Josep Carreras Leukaemia Foundation (content in Spanish)
Links of interest: local/provincial or state entities that can provide you with resources and services specialised in leukaemia or cancer patients:
In Spain there is a large network of associations for haematological cancer patients that, in many cases, can inform you, advise you and even carry out certain procedures. These are the contacts of some of them by Autonomous Communities:
All these organisations are external to the Josep Carreras Foundation.
STATE
- AEAL (ASOCIACIÓN ESPAÑOLA DE AFECTADOS POR LINFOMA, MIELOMA y LEUCEMIA)
- AECC (ASOCIACIÓN ESPAÑOLA CONTRA EL CÁNCER). Present in the different provinces and in many municipalities. Contact the nearest branch or call 900 100 036 (24h).
- AELCLES (Agrupación Española contra la Leucemia y Enfermedades de la Sangre)
- JOSEP CARRERAS LEUKAEMIA FOUNDATION
- FUNDACIÓN SANDRA IBARRA
- GEPAC (GRUPO ESPAÑOL DE PACIENTES CON CÁNCER)
ANDALUCÍA
- AECC (ASOCIACIÓN ESPAÑOLA CONTRA EL CÁNCER). Present in the different provinces and in many municipalities. Contact the nearest branch.
- ALUSVI (ASOCIACIÓN LUCHA Y SONRÍE POR LA VIDA). Sevilla
- APOLEU (ASOCIACIÓN DE APOYO A PACIENTES Y FAMILIARES DE LEUCEMIA). Cádiz
ARAGÓN
- AECC (ASOCIACIÓN ESPAÑOLA CONTRA EL CÁNCER). Present in the different provinces and in many municipalities. Contact the nearest branch.
- ASPHER (ASOCIACIÓN DE PACIENTES DE ENFERMEDADES HEMATOLÓGICAS RARAS DE ARAGÓN)
- DONA MÉDULA ARAGÓN
ASTURIAS
- AECC (ASOCIACIÓN ESPAÑOLA CONTRA EL CÁNCER). Present in the different provinces and in many municipalities. Contact the nearest branch.
- ASTHEHA (ASOCIACIÓN DE TRASPLANTADOS HEMATOPOYÉTICOS Y ENFERMOS HEMATOLÓGICOS DE ASTURIAS)
CANTABRIA
- AECC (ASOCIACIÓN ESPAÑOLA CONTRA EL CÁNCER). Present in the different provinces and in many municipalities. Contact the nearest branch.
CASTILLA LA MANCHA
- AECC (ASOCIACIÓN ESPAÑOLA CONTRA EL CÁNCER). Present in the different provinces and in many municipalities. Contact the nearest branch.
CASTILLA LEÓN
- ABACES (ASOCIACIÓN BERCIANA DE AYUDA CONTRA LAS ENFERMEDADES DE LA SANGRE)
- AECC (ASOCIACIÓN ESPAÑOLA CONTRA EL CÁNCER). Present in the different provinces and in many municipalities. Contact the nearest branch.
- ALCLES (ASOCIACIÓN LEONESA CON LAS ENFERMEDADES DE LA SANGRE). León.
- ASCOL (ASOCIACIÓN CONTRA LA LEUCEMIA Y ENFERMEDADES DE LA SANGRE). Salamanca.
CATALUÑA
- ASSOCIACIÓ FÈNIX. Solsona
- FECEC (FEDERACIÓ CATALANA D’ENTITATS CONTRA EL CÁNCER
- FUNDACIÓ KÁLIDA. Barcelona
- FUNDACIÓ ROSES CONTRA EL CÀNCER. Roses
- LLIGA CONTRA EL CÀNCER COMARQUES DE TARRAGONA I TERRES DE L’EBRE. Tarragona
- ONCOLLIGA BARCELONA. Barcelona
- ONCOLLIGA GIRONA. Girona
- ONCOLLIGA COMARQUES DE LLEIDA. Lleida
- ONCOVALLÈS. Vallès Oriental
- OSONA CONTRA EL CÀNCER. Osona
- SUPORT I COMPANYIA. Barcelona
- VILASSAR DE DALT CONTRA EL CÀNCER. Vilassar de Dalt
VALENCIAN COMMUNITY
- AECC (ASOCIACIÓN ESPAÑOLA CONTRA EL CÁNCER). Present in the different provinces and in many municipalities. Contact the nearest branch.
- ASLEUVAL (ASOCIACIÓN DE PACIENTES DE LEUCEMIA, LINFOMA, MIELOMA Y OTRAS ENFERMEDADES DE LA SANGRE DE VALENCIA)
EXTREMADURA
- AECC (ASOCIACIÓN ESPAÑOLA CONTRA EL CÁNCER). Present in the different provinces and in many municipalities. Contact the nearest branch.
- AFAL (AYUDA A FAMILIAS AFECTADAS DE LEUCEMIAS, LINFOMAS; MIELOMAS Y APLASIAS)
- AOEX (ASOCIACIÓN ONCOLÓGICA EXTREMEÑA)
GALICIA
- AECC (ASOCIACIÓN ESPAÑOLA CONTRA EL CÁNCER). Present in the different provinces and in many municipalities. Contact the nearest branch.
- ASOTRAME (ASOCIACIÓN GALLEGA DE AFECTADOS POR TRASPLANTES MEDULARES)
BALEARIC ISLANDS
- ADAA (ASSOCIACIÓ D’AJUDA A L’ACOMPANYAMENT DEL MALALT DE LES ILLES BALEARS)
- AECC (ASOCIACIÓN ESPAÑOLA CONTRA EL CÁNCER). Present in the different provinces and in many municipalities. Contact the nearest branch.
CANARY ISLANDS
- AECC (ASOCIACIÓN ESPAÑOLA CONTRA EL CÁNCER). Present in the different provinces and in many municipalities. Contact the nearest branch.
- AFOL (ASOCIACIÓN DE FAMILIAS ONCOHEMATOLÓGICAS DE LANZAROTE)
- FUNDACIÓN ALEJANDRO DA SILVA
LA RIOJA
- AECC (ASOCIACIÓN ESPAÑOLA CONTRA EL CÁNCER). Present in the different provinces and in many municipalities. Contact the nearest branch.
MADRID
- AECC (ASOCIACIÓN ESPAÑOLA CONTRA EL CÁNCER). Present in the different provinces and in many municipalities. Contact the nearest branch.
- AEAL (ASOCIACIÓN ESPAÑOLA DE LEUCEMIA Y LINFOMA)
- CRIS CONTRA EL CÁNCER
- FUNDACIÓN LEUCEMIA Y LINFOMA
MURCIA
- AECC (ASOCIACIÓN ESPAÑOLA CONTRA EL CÁNCER). Present in the different provinces and in many municipalities. Contact the nearest branch.
NAVARRA
- AECC (ASOCIACIÓN ESPAÑOLA CONTRA EL CÁNCER). Present in the different provinces and in many municipalities. Contact the nearest branch.
BASQUE COUNTRY
- AECC (ASOCIACIÓN ESPAÑOLA CONTRA EL CÁNCER). Present in the different provinces and in many municipalities. Contact the nearest branch.
- PAUSOZ-PAUSO. Bilbao
AUTONOMOUS CITIES OF CEUTA AND MELILLA
- AECC CEUTA (ASOCIACIÓN ESPAÑOLA CONTRA EL CÁNCER)
- AECC MELILLA (ASOCIACIÓN ESPAÑOLA CONTRA EL CÁNCER)
Support and assistance
We also invite you to follow us through our main social media (Facebook, Twitter and Instagram) where we often share testimonies of overcoming this disease.
If you live in Spain, you can also contact us by sending an e-mail to imparables@fcarreras.es so that we can help you get in touch with other people who have overcome this disease.