What is Fanconi Anaemia and who does it affect?
Fanconi anaemia is an inherited disease which is quite rare among the population. Despite its name, it is often characterised by many other manifestations in addition to anaemia.
The most relevant:
- Progressive bone marrow failure (anaemia, leukopaenia and thrombocytopaenia).
- Congenital malformations
- High predisposition to tumours, especially acute myeloblastic leukaemias (AML), myelodysplastic syndromes (MDS) and solid tumours.
 Its frequency is 1 in 300,000. It is a recessive genetic disease in most cases. Therefore, for an individual to have Fanconi anaemia, both copies of the genes involved in Fanconi anaemia (the one inherited from the father and the one inherited from the mother) must be affected. The condition is usually diagnosed in children between 3 and 14 years of age. When genes are implicated in tumour predisposition in carriers, appropriate follow-up in specialised units is important. Appropriate genetic counselling of at-risk relatives is essential, especially in carrier couples.
Its frequency is 1 in 300,000. It is a recessive genetic disease in most cases. Therefore, for an individual to have Fanconi anaemia, both copies of the genes involved in Fanconi anaemia (the one inherited from the father and the one inherited from the mother) must be affected. The condition is usually diagnosed in children between 3 and 14 years of age. When genes are implicated in tumour predisposition in carriers, appropriate follow-up in specialised units is important. Appropriate genetic counselling of at-risk relatives is essential, especially in carrier couples.
Genetic counselling
As most cases are recessive mutations, for an individual to have Fanconi anaemia, both alleles of a given FANC gene inherited from the parents must have the pathogenic mutation.
If a man and a woman, both carriers of mutations in a given FANC gene, have offspring, 3 out of 4 children, on average, will be healthy (on average, 2 will be carriers and 1 will be completely normal: neither of the two alleles of the gene will be affected).
However, on average, 1 in 4 offspring will have mutations in both alleles, and will therefore suffer from the disease with a severity that is difficult to define in advance. In the in the case of the FANCB gene, due to it being on the X chromosome, all those affected are male and only mothers are carriers. In these families it is very important to detect carrier females of reproductive age due to the high risk of conceiving an affected male child.
Individuals with one functional copy and one mutated copy are defined as carriers of the disease. Except in the case of carriers with a mutation in the FANCD1/BRCA2, FANCS/BRCA1, FANCN/PALB2 or FANCJ/BRIP1 genes, which affect a minority of patients, there is no evidence that FA carriers have a higher risk of disease than the rest of the population.
Other considerations within genetic counselling (family planning, prenatal diagnosis or pre-implantation diagnosis):
It is important to contact specialists so these can provide genetic counselling that will help a person or a family to understand the risk of a genetic disease, educate people about the disease, and assess the risk of transmitting the disease to their children, to have proper family planning and correct prenatal or pre-implantation diagnosis.
The genetic counsellor often works with families to identify individuals at risk. If appropriate or necessary, this counsellor will discuss genetic testing, interpret results, and oversee any additional tests, research aids or options available to family members. Genetic counsellors work as part of a health team, together with doctors, social workers, nurses, clinical geneticists and other specialists to help families make informed decisions about their health. They will also be a source of help and support in difficult times.
If you wish to contact specialists who can help you in the diagnosis, follow-up and treatment of patients with Fanconi anaemia, you can do so in the following way:
Fanconi Anaemia Foundation Spain
secretariaFAF@anemiadefanconi.org
654 617 808
What are the causes of Fanconi anaemia?
 Fanconi anaemia is a complex disease from a genetic point of view, as 22 genes involved in this disease have been described so far. The lack of function of any of these 22 genes results in the disease. These abnormal genes damage cells, preventing them from repairing damaged DNA. The disease is mainly due to mutations in genes, and there are some correlations between Fanconi anaemia subtypes, genetic mutations and disease behaviour.
Fanconi anaemia is a complex disease from a genetic point of view, as 22 genes involved in this disease have been described so far. The lack of function of any of these 22 genes results in the disease. These abnormal genes damage cells, preventing them from repairing damaged DNA. The disease is mainly due to mutations in genes, and there are some correlations between Fanconi anaemia subtypes, genetic mutations and disease behaviour.
In patients with FANC C mutations, aplasia is earlier and survival is shorter than in carriers of FANC A and FANC G mutations.
Patients with FANC G have a higher risk of developing leukaemias and solid tumours than those with FANC A type.
FANC D1 and FANC N subtypes carry a risk of developing medulloblastomas or Wilms tumours. Heterozygous mutations in FANC D1, FANC J and FANC N carry an increased risk of breast cancer.
What are the symptoms of Fanconi anaemia?
Approximately 90% of children with Fanconi anaemia have alterations in bone marrow function leading to aplastic anaemia. Affected individuals suffer from fatigue and weakness due to anaemia, recurrent infections due to neutropaenia and clotting problems due to thrombocytopaenia.
The clinical features associated with Fanconi anaemia are pre- and postnatal growth retardation, renal, gastrointestinal, genitourinary, cardiac and skeletal malformations, small head, eyes and mouth, hypogonadism, partial deafness, skin abnormalities such as hyper- or hypopigmentation and café-au-lait spots and elevated a-fetoprotein levels in the blood. Three out of ten patients, however, have no malformation at all.
Haematological problems usually appear at school age, around the age of 7, although this is highly variable. Haematological disorders affect most Fanconi anaemia patients before the age of 40. 90% of cases are diagnosed before adolescence. Patients show abnormally low blood cell counts, both of red blood cells (anaemia), white blood cells (leukopaenia) and of platelets (thrombopaenia). The first manifestation is usually isolated thrombopaenia in more than 50% of cases, with petechiae or haematomas, or episodes of nasal or gastrointestinal bleeding. Subsequently, signs of anaemia become evident, including mainly pallor, asthaenia and anorexia. The tendency to develop infectious processes (secondary to white blood cell deficiency) is usually of late onset. Once haematological involvement has begun, the course usually leads to pancytopaenia (anaemia + leukopaenia + thrombopaenia) within an extraordinarily variable period of time.
How is Fanconi anaemia diagnosed?
- Clinical diagnosis
Diagnosis based on clinical observations can be extremely difficult due to the great variability of symptoms displayed by Fanconi anaemia patients. This fact, together with the fact that 30% of FA patients do not display any malformation, means that many Fanconi anaemia patients only see a doctor when the symptoms of the haematological disease have manifested themselves. A significant reduction in one or more blood series usually raises the specialist’s suspicion of hereditary anaemia, which must be confirmed by additional clinical studies as well as cytogenetic and molecular analyses.
- Genetic diagnosis
Fanconi anaemia is a complex disease from a genetic point of view, as 22 genes involved in this disease have so far been described: FANCA, FANCB, FANCC, FANCD1/BRCA2, FANCD2, FANCE, FANCF, FANCG/XRCC9, FANCI, FANCJ/BRIP1/BACH1, FANCL, FANCM, FANCN/PALB2, FANCO/RAD51C, FANCP/SLX4/BTBD12, FANCQ/ERCC4/XPF, FANCR/RAD51, FANCS/BRCA1, FANCT/UBE2T, FANCU/XRCC2, FANCV/REV7, FANCW/RFWD3
Diagnostic confirmation of the disease is carried out by testing for chromosomal fragility (chromosome breaks) induced by agents that generate cross-links in the DNA strands. These studies are usually performed on peripheral blood lymphocytes or skin fibroblasts. These tests must be performed by laboratories experienced in the testing of genetic diseases. The National Fanconi Anaemia Research Network offers Spanish patients diagnostic confirmations of the disease, as well as training for laboratories wishing to specialise in these tests. In approximately 20% of Fanconi anaemia patients, diagnostic confirmation of the disease can be particularly complex due to a phenomenon known as somatic mosaicism. Somatic mosaicism derives from the fact that some blood cells can correct, by different mechanisms, the mutation of the disease-causing gene, thus Fanconi anaemia cells and healthy cells coexist in the patient’s blood.
Diagnosis of the genetic subtype is carried out via sequencing techniques (Sanger or NGS) and MLPA. A mutational study can be performed to identify carriers of the disease, and to carry out prenatal or pre-implantation diagnosis studies.
- Differential diagnosis
It is important that the team has experience in the management of Fanconi anaemia patients. There are some diseases that can be confused in the diagnosis. To be considered:
-
- Dyskeratosis congenita
- Blacfan-Diamond anaemia
- Shwachman-Diamond syndrome
- Severe congenital neutropaenia
- TAR syndrome (thrombocytopaenia with absence of rays)
- Amegakaryocytic thrombocytopaenia
- Baller-Gerold syndrome
- Hijmegen breakage syndrome
- Rothmund-Thomson syndrome
- Roberts syndrome
- Warsaw breakage syndrome
- DK Phocomelia syndrome
- VACTERL with hydrocephalus
- Wiskott-Aldrich syndrome
What is the treatment for Fanconi anaemia?
Red blood cell and platelet transfusions are necessary when anaemia and thrombocytopaenia are severe and symptomatic.

Androgen derivatives (oxymetholone at doses of 2-4 mg/kg/day) may decrease the degree of anaemia and avoid the need for transfusions, but the occurrence of side effects (virilization, growth acceleration, liver dysfunction and liver tumours) should be monitored. Its use should be transitional while awaiting a haematopoietic stem cell transplant (bone marrow transplantation).
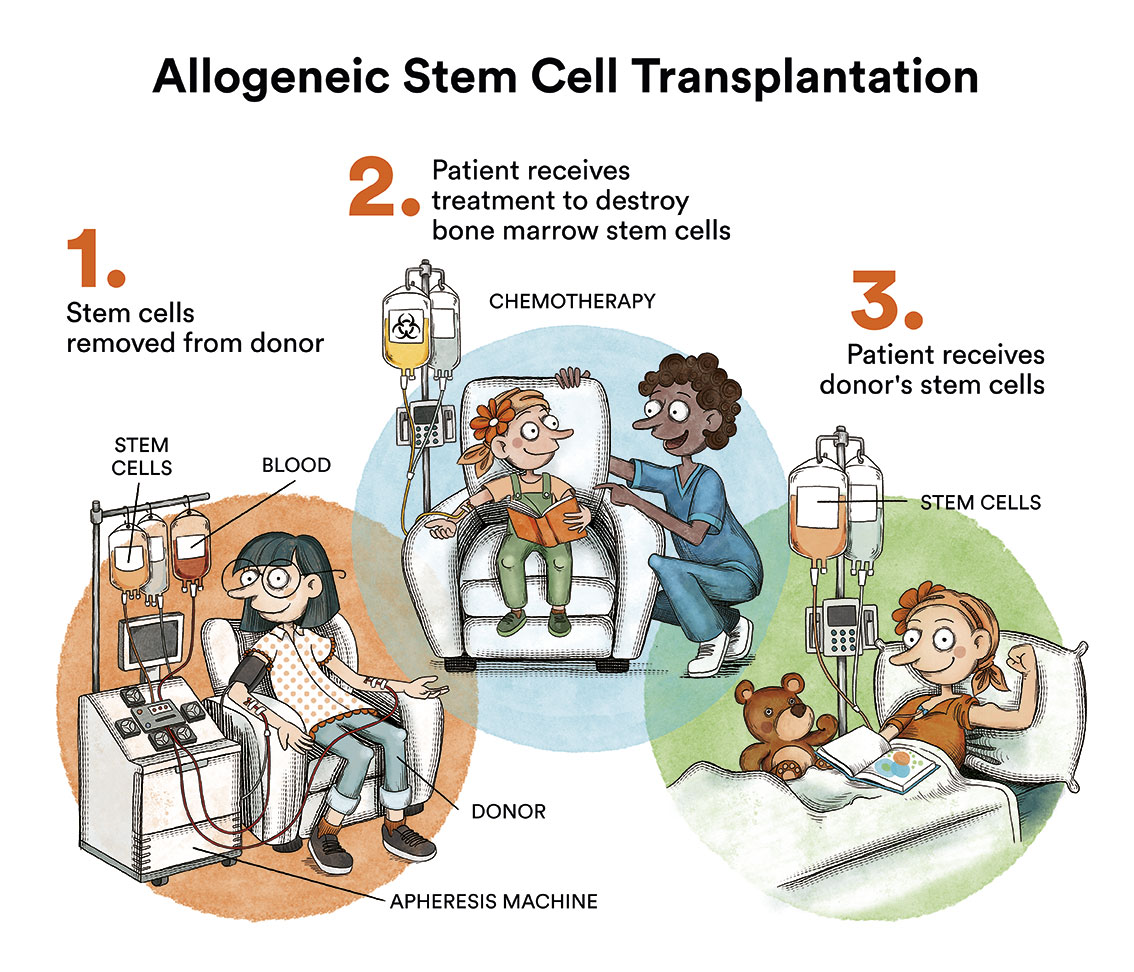
Haematopoietic stem cell transplantation is the only treatment that can restore normal haematopoiesis, but it does not reverse somatic lesions nor does it prevent the development of solid tumours. The ideal donor is an HLA-identical sibling, but currently, if this option is not available, transplants from HLA-compatible volunteer donors are also a very good option.
The use of classic myeloablative regimens with cyclophosphamide and irradiation lead to poor results due to the hypersensitivity of Fanconi anaemia cells to genotoxic agents, which cause severe tissue damage. Therefore, reduced-intensity conditioning regimens adapted to these patients should be used and radiotherapy should always be avoided.
Patients with Fanconi anaemia receiving a bone marrow transplant require close monitoring due to the risk of developing malignancies, including growth and endocrine check-ups, bone marrow cytogenetics and regular oral examinations with biopsy when any suspicious lesions appear.
What are the chances of chidren with Fanconi anaemia being cured?
 The likelihood of transformation of the disease in acute myeloid leukaemia is 30% and the incidence of solid tumours is 28% by the age of 40.
The likelihood of transformation of the disease in acute myeloid leukaemia is 30% and the incidence of solid tumours is 28% by the age of 40.
Without bone marrow transplantation, the median survival time for patients with Fanconi anaemia is 24 years. The incidence of spinal cord failure is as high as 90% by the age of 40. Death, in 90% of cases, is due to haematological disorders (bone marrow aplasia, myelodysplastic syndromes or acute myeloid leukaemia) or complications arising from treatment.
Transplants from a healthy, compatible sibling offer a chance of survival free of haematological disease of up to 90%. The use of an anonymous HLA donor is not optimal, but is improving every day. The prognosis associated with transplantation is better if the patient has received few transfusions, if the patient is young and if they have not developed a leukaemic process. Therefore, if the patient has a suitable donor, it is advisable to transplant before the patient starts to require transfusion support, or when bone marrow cytogenetic studies suggest the onset of myelodysplasia. Hence the importance of the correct monitoring of these patients in terms of their peripheral blood and bone marrow. The National Research Network on Fanconi Anaemia has standardised protocols for the transplantation of these patients according to the best international standards, and provides information to specialised centres that may request it. Contact the Fanconi Anaemia Foundation.
Monitoring
- Haematological monitoring
Patients with Fanconi anaemia are at high risk of developing aplastic anaemia, myelodysplastic syndromes or acute myeloid leukaemia. The likelihood of transformation of the disease into acute myeloid leukaemia is 30% and the incidence of solid tumours is 28% by the age of 40. It is very important to closely monitor their haematological parameters. As a guideline, the recommended controls are as follows:
-
- Haemogram via peripheral blood sampling every 3-4 months.
- Myelogram, cytogenetics, CD34+ analysis and CFCs content) by bone marrow aspirate every 12 to 18 months if no significant changes are observed.
Haematological monitoring of the patient should begin as soon as the disease is diagnosed.
- Monitoring and prevention of head and neck tumours
Patients with Fanconi anaemia have a much higher predisposition than the rest of the population to develop solid tumours. For this reason, monitoring by specialists
who have in-depth knowledge of the disease is of vital importance. There is a cumulative risk of developing some kind of tumour at 40 years of age of 25%. No cases have been reported in patients under 10 years of age. In transplant recipients the risk increases from the 5th year post-transplant. The recommendations for routine check-ups and monitoring are as follows:
-
- Non-transplanted patients: from 10 years of age
- Transplant recipients: from the time of transplantation regardless of age
Monitoring of the oral cavity, nasopharynx, oropharynx, hypopharynx and larynx should be established every six months if there are no significant changes. In case of changes, lichenplanus, leukoplakia and erythroplakia should be monitored via biopsy every 2-3 months. And if there is a lesion suspicious of being carcinoma, a biopsy should be taken immediately and the patient should be monitored every 2-3 months, in addition to an annual X-ray.
- Monitoring and prevention of gynaecological tumours
Because of the risk of cervical, vaginal and vulvar cancer, gynaecological monitoring should begin at age 16 or at the first menstrual period. In the same way, a breast examination should also be carried out from the first menstruation or from the age of 20.
Monitoring should consist of an annual Pap smear, thorough examination of the lower genital tract, endocrine monitoring and papillomavirus testing.
- Dental care and monitoring
It is recommended to start dental check-ups at the age of one and a half years and to perform 2 check-ups per year. It is recommended that the dentist treating a Fanconi anaemia patient be put in contact with the patient’s specialist physician in order to be informed about possible complications associated with oral interventions.
It is recommended to check for persistent lesions, suspicious ulcerations or leukoplakia and to contact the specialist in case an oral tissue biopsy is required. It is also recommended to contact the specialist in the case of continuous bleeding or tooth loss without apparent cause.
If the patient has abnormally low levels of platelets or white blood cells, the dentist should be informed in order to prevent bleeding or infection if the patient is going to undergo dental surgery.
Links of interest on other topics related to Fanconi anaemia
BONE MARROW TRANSPLANT
- Bone Marrow Transplant Guide. Josep Carreras Foundation (content in Spanish)
- What is HLA and how does it work? Josep Carreras Foundation (content in Spanish)
- Graft-versus-Host Disease. Josep Carreras Foundation (content in Spanish)
- History of Bone Marrow Transplantation. Josep Carreras Foundation (content in Spanish)
- How is the search for an anonymous donor conducted? Josep Carreras Foundation (content in Spanish)
Useful links: local entities (resources and services)
All these organisations are external to the Josep Carreras Foundation.
ANDALUCÍA
ARAGÓN
ASTURIAS
CASTILLA LA MANCHA
CASTILLA LEÓN
CATALUÑA
VALENCIAN COMMUNITY
EXTREMADURA
GALICIA
BALEARIC ISLANDS
CANARY ISLANDS
LA RIOJA
MADRID
- AAA (asociación de adolescentes y Adultos Jóvenes con Cáncer)
- ASION
- FUNDACIÓN CAICO
- FUNDACIÓN ALADINA
- FUNDACIÓN UNOENTRECIENMIL
MURCIA
NAVARRA
BASQUE COUNTRY
Support and assistance
We also invite you to follow us through our main social media (Facebook, Twitter and Instagram) where we often share testimonies of overcoming this disease.
If you live in Spain, you can also contact us by sending an e-mail to imparables@fcarreras.es so that we can help you get in touch with other people who have overcome this disease.