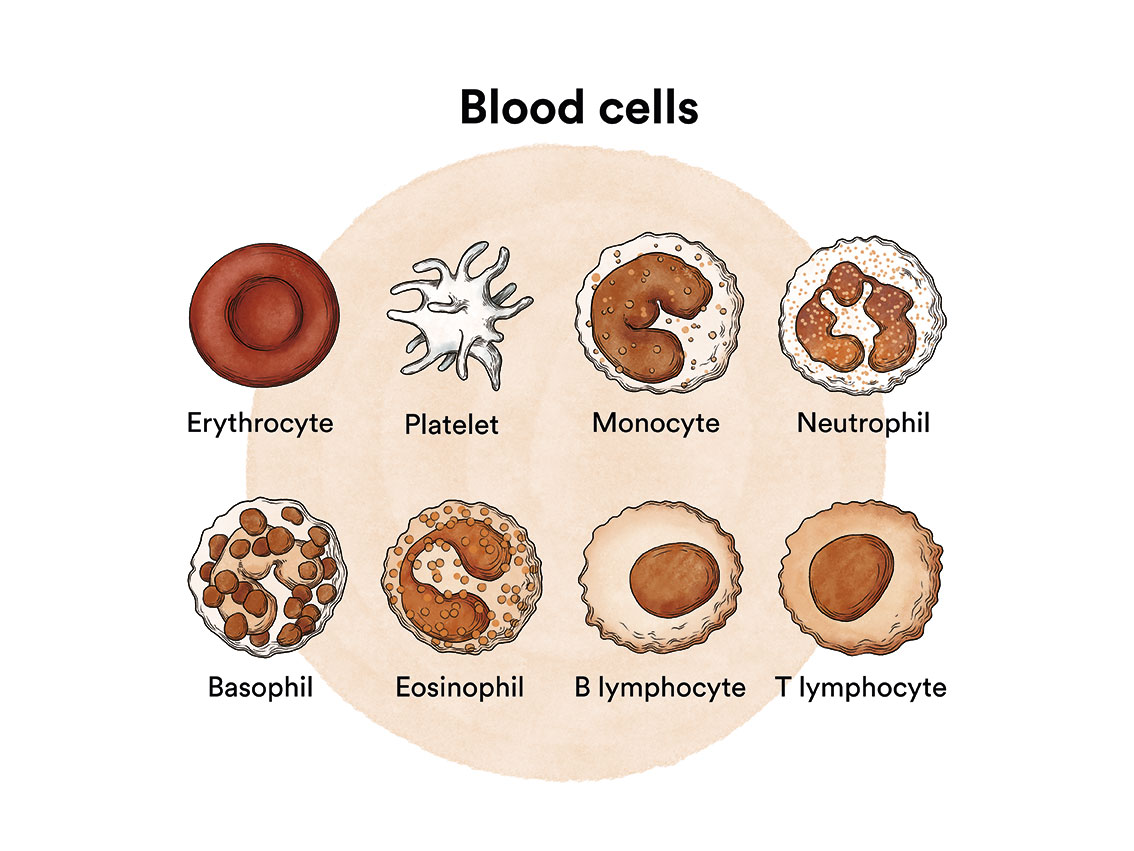
How does bone marrow work and what are the types of blood cells?
Primary myelofibrosis, also known as idiopathic myelofibrosis or agnogenic myeloid metaplasia, is a type of blood cell cancer that originates in the bone marrow. See section Leukaemia, bone marrow and blood cells.
What is primary myelofibrosis and who does it affect?
Myelofibrosis is a malignant disease that falls into the group of chronic myeloproliferative neoplasms, along with polycythaemia vera and essential thrombocythaemia, among others.
 In these diseases, the bone marrow stem cells, which are responsible for making all blood cells, have a defect that causes them to produce some of the myeloid blood cells in an uncontrolled manner. This genetic disorder is not inherited (not passed on from parents to children), although some families are predisposed to develop myeloproliferative neoplasms.
In these diseases, the bone marrow stem cells, which are responsible for making all blood cells, have a defect that causes them to produce some of the myeloid blood cells in an uncontrolled manner. This genetic disorder is not inherited (not passed on from parents to children), although some families are predisposed to develop myeloproliferative neoplasms.
Primary myelofibrosis is characterised by the presence of fibrous tissue in the bone marrow and a shift of stem cells from the bone marrow into the blood, where they colonise distant organs (primarily the spleen and liver).
It is a very rare disease. Its incidence in Spain is around 5-7 cases per million inhabitants per year. It predominates in older patients, with an average age at diagnosis of 65 years. It is more common in males and is very occasionally diagnosed in childhood.
A proportion of patients with polycythaemia vera and essential thrombocythaemia will eventually develop marrow fibrosis, indistinguishable from primary myelofibrosis.
We recommend reading the Educational guide on myelofibrosis of the Spanish Group of Philadelphia Negative Chronic Myeloproliferative Diseases (GEMFIN) of the Spanish Society of Haematology and Haemotherapy (SEHH).
What are the causes of primary myelofibrosis?
It is not known why primary myelofibrosis occurs.
At the cytogenetic level, primary myelofibrosis is caused by genetic mutations. These mutations alter the system that regulates the formation of blood cells, known as the JAK-STAT pathway. In people with myelofibrosis , this pathway is permanently activated, i.e. it does not stop working at any time, causing the stem cells to keep multiplying and, moreover, to function in an altered way.
What are the symptoms of primary myelofibrosis?
In one third of cases, the disease has a symptomless onset and is therefore detected by chance in a routine blood test. When clinical manifestations are present, they usually include: constitutional symptoms (30% of cases): lack of appetite, weight loss, profuse sweating in the evening, febrile fever; anaemic syndrome (25% of cases): tiredness, dyspnoea (shortness of breath) on exertion, oedema in the legs; abdominal discomfort related to enlargement of the spleen (splenomegaly) (20% of cases); bloating after meals, pain in the left side of the abdomen; or, less frequently, arterial or venous thrombosis (7% of cases), bleeding, recurrent infections, generalised pruritus (itching), bone pain or gout crises (due to hyperuricaemia).
How is primary myelofibrosis diagnosed?
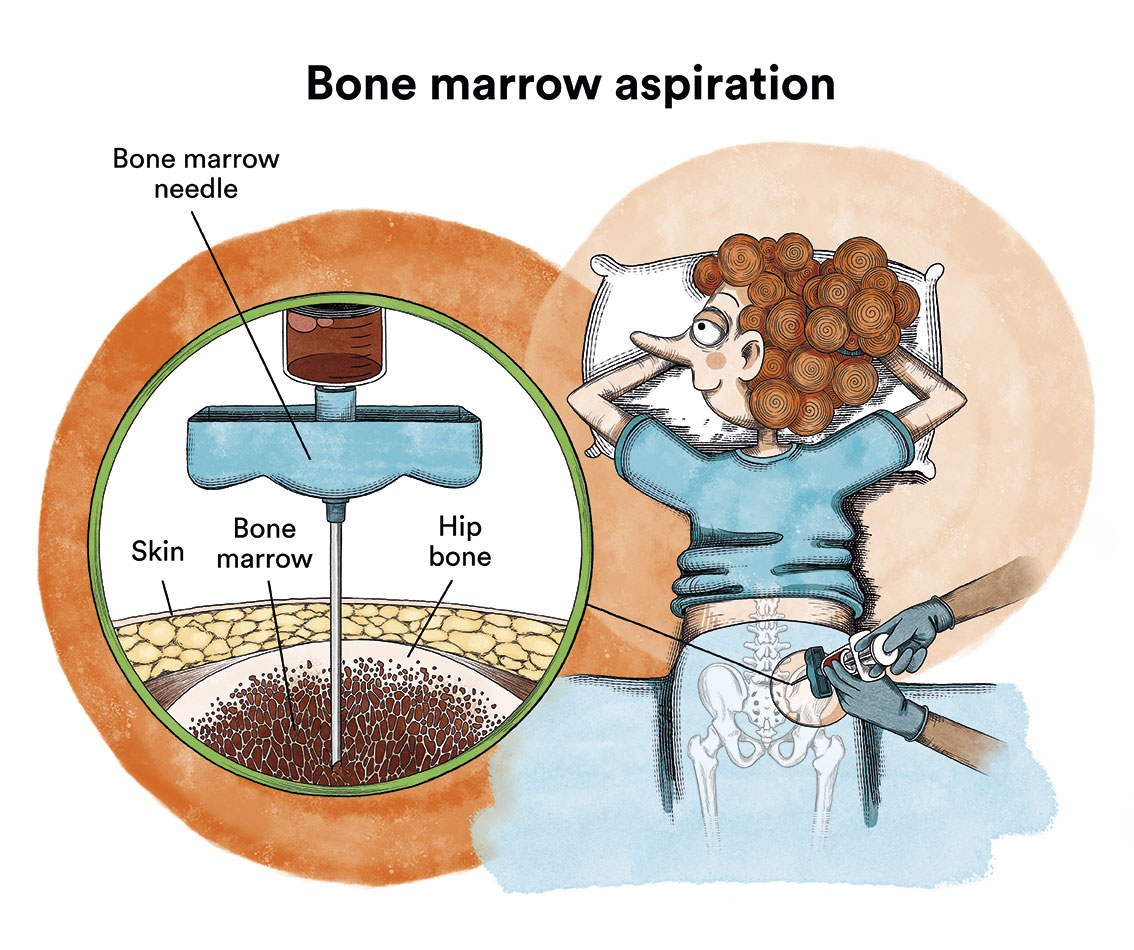
It is not possible to confirm the diagnosis of myelofibrosis with a blood test alone. A bone marrow biopsy and blood tests are essential for diagnosis. Cytogenetic and molecular studies should also be carried out, which are important both diagnostically and for estimating the prognosis of the disease. In this respect, more than two thirds of patients will have a mutation in one or more of the following genes: JAK2 (50-60%), CALR (20-30%) or MPL (5-10%), which is present in their blood cells and is a marker for the disease.
What is the treatment for primary myelofibrosis?
 Myelofibrosis is a very clinically heterogeneous disease that requires individualised risk-adjusted treatment. Some people can live symptom-free for many years. Others, on the other hand, may have an aggressive disease from the outset or one that becomes progressively worse. In both cases, patients should be monitored regularly at their referral centre.
Myelofibrosis is a very clinically heterogeneous disease that requires individualised risk-adjusted treatment. Some people can live symptom-free for many years. Others, on the other hand, may have an aggressive disease from the outset or one that becomes progressively worse. In both cases, patients should be monitored regularly at their referral centre.
The first decision regarding the management of a patient with myelofibrosis is whether or not treatment is required. If the patient is asymptomatic and has no laboratory data that could pose a potential risk, it is feasible to maintain a watchful waiting approach and perform regular monitoring with a view to initiating treatment when necessary.
If not, it must be determined whether the patient is a candidate for an allogeneic haematopoietic stem cell transplant (from a related or unrelated donor). There are different survival prognoses based on variables such as age, constitutional symptoms (weight loss, fever, etc.), laboratory results and cytogenetic and molecular characteristics of the disease itself, which classify it from low risk to high risk and serve as a guide for treatment.
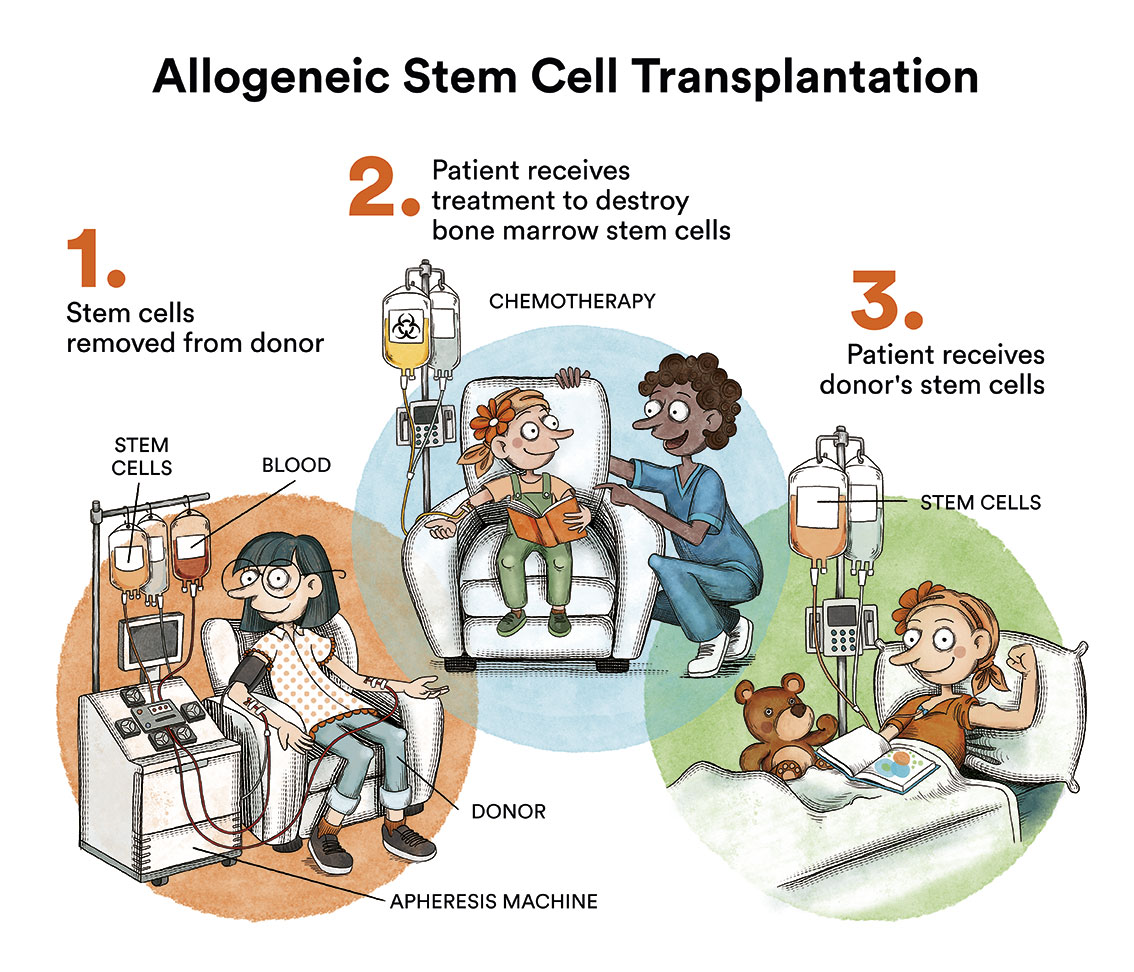
Indeed, allogeneic transplantation is the only curative treatment for myelofibrosis at present. However, since it carries a significant mortality and morbidity, this procedure should be reserved for young patients in good general condition with poor prognosis myelofibrosis (high risk according to prognostic indices).
In practice, the vast majority of patients are not candidates for transplantation and their treatment is aimed at symptom control. For this purpose, different therapeutic strategies are available, generally aimed at improving anaemia or controlling hyperproliferative manifestations (constitutional symptoms and painful splenomegaly). In this regard, the most notable advance in recent years has been the introduction of ruxolitinib, a drug that is highly effective in controlling the hyperproliferative manifestations of the disease and pruritus, which is usually accompanied by a substantial improvement in the patient’s quality of life.
When these symptoms are not very pronounced, hydroxyurea can be used, due to its easy handling and good tolerance. Erythropoietic agents (erythropoietin, darbepoetin) and danazol are particularly effective in improving anaemia, while immunomodulatory drugs (thalidomide, lenalidomide) or low-dose corticosteroids may be useful in cases refractory to treatment. Splenectomy (surgical removal of the spleen) has been virtually abandoned due to its high morbidity and mortality, although it may be indicated in very selected cases. The available therapeutic options have transient efficacy, thus the inclusion of patients in clinical trials with new drugs should be considered.
What is the prognosis for patients with primary myelofibrosis?
Survival of patients with myelofibrosis varies greatly depending on the presence or absence of unfavourable prognostic factors (advanced age, severe anaemia, constitutional symptoms, leukocytosis, platelets, circulating blasts, high-risk cytogenetic or molecular alterations).
The main causes of death are progressive clinical deterioration resulting from the myelofibrosis itself, leukaemic transformation (20% within 10 years of diagnosis), heart failure, infections, thrombotic and haemorrhagic complications.
We advise myelofibrosis patients in Spain to contact MPN, the Spanish association of patients with chronic myeloproliferative neoplasms.
Links of interest concerning medical issues relating to primary myelofibrosis
Primary myelofibrosis. The Leukaemia Foundation
Myelofibrosis (MF). Leukemia & Lymphoma Society
Myelofibrosis (MF). Bloodwise UK
Links of interest on other topics related to primary myelofibrosis
TESTIMONIAL MATERIALS
You can order the booklets in paper format for free delivery in Spain by e-mail: imparables@fcarreras.es
BONE MARROW TRANSPLANT
- Bone Marrow Transplant Guide. Josep Carreras Foundation (content in Spanish)
- What is HLA and how does it work? Josep Carreras Foundation (content in Spanish)
- Graft-versus-Host Disease. Josep Carreras Foundation (content in Spanish)
- History of Bone Marrow Transplantation. Josep Carreras Foundation (content in Spanish)
- How is the search for an anonymous donor conducted? Josep Carreras Foundation (content in Spanish)
FOOD
- How to maintain a healthy diet during treatment? Josep Carreras Foundation (content in Spanish)
- Nutrition guide. Leukemia & Lymphoma Society
OTHER
- Ideas on what to take with me to the isolation chamber. Josep Carreras Leukaemia Foundation (content in Spanish)
- Travel tips for people with cancer. Josep Carreras Leukaemia Foundation (content in Spanish)
- Physiotherapy manual for haematological and transplant patients. Josep Carreras Leukaemia Foundation (content in Spanish)
- Prevention and treatment of oral mucositis. Josep Carreras Leukaemia Foundation (content in Spanish)
- Oral hygiene in oncohaematological patients. Josep Carreras Leukaemia Foundation (content in Spanish)
- Fertility manual: Suffering from blood cancer and becoming a parent. Josep Carreras Leukaemia Foundation (content in Spanish)
- Skin care in the oncohaematological patient. Josep Carreras Leukaemia Foundation (content in Spanish)
- Aesthetic Oncology Manual. Josep Carreras Leukaemia Foundation (content in Spanish)
- Leukaemia and sexuality. Josep Carreras Leukaemia Foundation (content in Spanish)
- 7 ways to wear a scarf. Josep Carreras Leukaemia Foundation (content in Spanish)
Links of interest: local/provincial or state entities that can provide you with resources and services specialised in leukaemia or cancer patients:
In Spain there is a large network of associations for haematological cancer patients that, in many cases, can inform you, advise you and even carry out certain procedures. These are the contacts of some of them by Autonomous Communities:
All these organisations are external to the Josep Carreras Foundation.
STATE
- MPN España (Asociación de Afectados Por Neoplasias Mieloproliferativas Crónicas)
- CEMMP (Comunidad Española de Pacientes de Mieloma Múltiple)
- AEAL (ASOCIACIÓN ESPAÑOLA DE AFECTADOS POR LINFOMA, MIELOMA y LEUCEMIA)
- AECC (ASOCIACIÓN ESPAÑOLA CONTRA EL CÁNCER). Present is the different provinces and in many municipalities. Contact with the nearest branch or call 900 100 036 (24h).
- AELCLES (Agrupación Española contra la Leucemia y Enfermedades de la Sangre)
- Josep Carreras Leukaemia Foundation
- FUNDACIÓN SANDRA IBARRA
- GEPAC (GRUPO ESPAÑOL DE PACIENTES CON CÁNCER)
ANDALUCÍA
- AECC (ASOCIACIÓN ESPAÑOLA CONTRA EL CÁNCER). Present is the different provinces and in many municipalities. Contact the nearest branch.
- ALUSVI (ASOCIACIÓN LUCHA Y SONRÍE POR LA VIDA). Sevilla
- APOLEU (ASOCIACIÓN DE APOYO A PACIENTES Y FAMILIARES DE LEUCEMIA). Cádiz
ARAGÓN
- AECC (ASOCIACIÓN ESPAÑOLA CONTRA EL CÁNCER). Present is the different provinces and in many municipalities. Contact the nearest branch.
- ASPHER (ASOCIACIÓN DE PACIENTES DE ENFERMEDADES HEMATOLÓGICAS RARAS DE ARAGÓN)
- DONA MÉDULA ARAGÓN
ASTURIAS
- AECC (ASOCIACIÓN ESPAÑOLA CONTRA EL CÁNCER). Present is the different provinces and in many municipalities. Contact the nearest branch.
- ASTHEHA (ASOCIACIÓN DE TRASPLANTADOS HEMATOPOYÉTICOS Y ENFERMOS HEMATOLÓGICOS DE ASTURIAS)
CANTABRIA
- AECC (ASOCIACIÓN ESPAÑOLA CONTRA EL CÁNCER). Present is the different provinces and in many municipalities. Contact the nearest branch.
CASTILLA LA MANCHA
- AECC (ASOCIACIÓN ESPAÑOLA CONTRA EL CÁNCER). Present is the different provinces and in many municipalities. Contact the nearest branch.
CASTILLA LEÓN
- ABACES (ASOCIACIÓN BERCIANA DE AYUDA CONTRA LAS ENFERMEDADES DE LA SANGRE)
- AECC (ASOCIACIÓN ESPAÑOLA CONTRA EL CÁNCER). Present is the different provinces and in many municipalities. Contact the nearest branch.
- ALCLES (ASOCIACIÓN LEONESA CON LAS ENFERMEDADES DE LA SANGRE). León.
- ASCOL (ASOCIACIÓN CONTRA LA LEUCEMIA Y ENFERMEDADES DE LA SANGRE). Salamanca.
CATALUÑA
- ASSOCIACIÓ FÈNIX. Solsona
- FECEC (FEDERACIÓ CATALANA D’ENTITATS CONTRA EL CÁNCER
- FUNDACIÓ KÁLIDA. Barcelona
- FUNDACIÓ ROSES CONTRA EL CÀNCER. Roses
- LLIGA CONTRA EL CÀNCER COMARQUES DE TARRAGONA I TERRES DE L’EBRE. Tarragona
- MielomaCAT
- ONCOLLIGA BARCELONA. Barcelona
- ONCOLLIGA GIRONA. Girona
- ONCOLLIGA COMARQUES DE LLEIDA. Lleida
- ONCOVALLÈS. Vallès Oriental
- OSONA CONTRA EL CÀNCER. Osona
- SUPORT I COMPANYIA. Barcelona
- VILASSAR DE DALT CONTRA EL CÀNCER. Vilassar de Dalt
VALENCIAN COMMUNITY
- AECC (ASOCIACIÓN ESPAÑOLA CONTRA EL CÁNCER). Present is the different provinces and in many municipalities. Contact the nearest branch.
- ASLEUVAL (ASOCIACIÓN DE PACIENTES DE LEUCEMIA, LINFOMA, MIELOMA Y OTRAS ENFERMEDADES DE LA SANGRE DE VALENCIA)
EXTREMADURA
- AECC (ASOCIACIÓN ESPAÑOLA CONTRA EL CÁNCER). Present is the different provinces and in many municipalities. Contact the nearest branch.
- AFAL (AYUDA A FAMILIAS AFECTADAS DE LEUCEMIAS, LINFOMAS; MIELOMAS Y APLASIAS)
- AOEX (ASOCIACIÓN ONCOLÓGICA EXTREMEÑA)
GALICIA
- AECC (ASOCIACIÓN ESPAÑOLA CONTRA EL CÁNCER). Present is the different provinces and in many municipalities. Contact the nearest branch.
- ASOTRAME (ASOCIACIÓN GALLEGA DE AFECTADOS POR TRASPLANTES MEDULARES)
BALEARIC ISLANDS
- ADAA (ASSOCIACIÓ D’AJUDA A L’ACOMPANYAMENT DEL MALALT DE LES ILLES BALEARS)
- AECC (ASOCIACIÓN ESPAÑOLA CONTRA EL CÁNCER). Present is the different provinces and in many municipalities. Contact the nearest branch.
CANARY ISLANDS
- AECC (ASOCIACIÓN ESPAÑOLA CONTRA EL CÁNCER). Present is the different provinces and in many municipalities. Contact the nearest branch.
- AFOL (ASOCIACIÓN DE FAMILIAS ONCOHEMATOLÓGICAS DE LANZAROTE)
- FUNDACIÓN ALEJANDRO DA SILVA
LA RIOJA
- AECC (ASOCIACIÓN ESPAÑOLA CONTRA EL CÁNCER). Present is the different provinces and in many municipalities. Contact the nearest branch.
MADRID
- AECC (ASOCIACIÓN ESPAÑOLA CONTRA EL CÁNCER). Present is the different provinces and in many municipalities. Contact the nearest branch.
- AEAL (ASOCIACIÓN ESPAÑOLA DE LEUCEMIA Y LINFOMA)
- CRIS CONTRA EL CÁNCER
- FUNDACIÓN LEUCEMIA Y LINFOMA
MURCIA
- AECC (ASOCIACIÓN ESPAÑOLA CONTRA EL CÁNCER). Present is the different provinces and in many municipalities. Contact the nearest branch.
NAVARRA
- AECC (ASOCIACIÓN ESPAÑOLA CONTRA EL CÁNCER). Present is the different provinces and in many municipalities. Contact the nearest branch.
BASQUE COUNTRY
- AECC (ASOCIACIÓN ESPAÑOLA CONTRA EL CÁNCER). Present is the different provinces and in many municipalities. Contact the nearest branch.
- PAUSOZ-PAUSO. Bilbao
AUTONOMOUS CITIES OF CEUTA AND MELILLA
- AECC CEUTA (ASOCIACIÓN ESPAÑOLA CONTRA EL CÁNCER)
- AECC MELILLA (ASOCIACIÓN ESPAÑOLA CONTRA EL CÁNCER)
Support and assistance
We also invite you to follow us through our main social media (Facebook, Twitter and Instagram) where we often share testimonies of overcoming this disease.
If you live in Spain, you can also contact us by sending an e-mail to imparables@fcarreras.es so that we can help you get in touch with other people who have overcome this disease.
