
Informació avalada per… ![]()
Què és la leucèmia, la medul·la òssia i quins són els tipus de cèl·lules sanguínies?
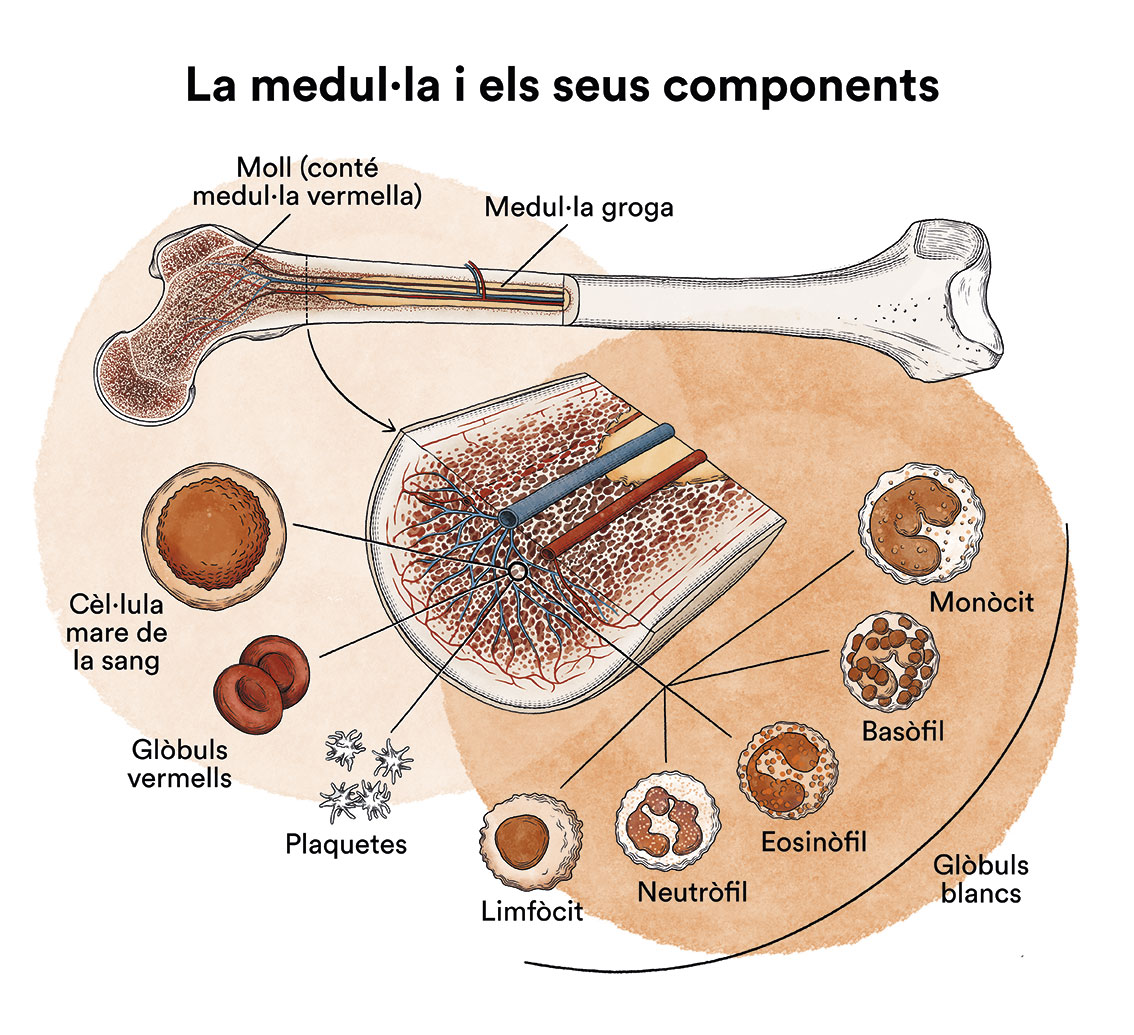
La leucèmia és un tipus de càncer de les cèl·lules de la sang i de la medul·la òssia. Vegeu apartat Leucèmia, medul·la òssia i cèl·lules sanguínies.
Què és la leucèmia mieloide aguda infantil?
 Les leucèmies agudes són un grup de malalties neoplàstiques caracteritzades per la transformació maligna i producció incontrolada de cèl·lules hematopoètiques immadures de la línia limfoide (leucèmia limfoblàstica aguda, LLA) o mieloide (leucèmia mieloblàstica aguda, LMA).
Les leucèmies agudes són un grup de malalties neoplàstiques caracteritzades per la transformació maligna i producció incontrolada de cèl·lules hematopoètiques immadures de la línia limfoide (leucèmia limfoblàstica aguda, LLA) o mieloide (leucèmia mieloblàstica aguda, LMA).
En la leucèmia mieloide aguda, les cèl·lules immadures de la línia mieloide (mieloblasts) proliferen de forma anormal i envaeixen progressivament la medul·la òssia i interfereixen en la producció de cèl·lules normals de la sang, cosa que origina insuficiència medul·lar i infiltra teixits extramedul·lars.
La LMA constitueix un 20 % de les leucèmies diagnosticades en aquesta etapa de la vida. La incidència anual en l’edat pediàtrica de la LMA és de 8 casos per cada milió de nens i nenes de menys de 15 anys (US Cancer Institute ‘s Surveillance, Epidemiology and End Results (SEER) program).
La LMA en el període infantil és més freqüent abans dels dos anys.
La seva incidència en l’etapa escolar descendeix i augmenta progressivament amb l’edat a partir de l’adolescència.
Com hem vist a “Leucèmia, medul·la òssia i cèl·lules sanguínies” , la medul·la òssia elabora les cèl·lules mare sanguínies (cèl·lules immadures) que, amb el temps, es transformaran en cèl·lules sanguínies madures. Una cèl·lula mare sanguínia es converteix en una cèl·lula mare mieloide o en una cèl·lula mare limfoide.
Una cèl·lula mare mieloide es converteix en un dels tres tipus de glòbuls sanguinis madurs:
- Glòbuls vermells, que transporten l’oxigen a altres tipus i òrgans del cos
- Granulòcits, glòbuls blancs que ajuden a combatre infeccions i altres malalties
- Plaquetes, que col·laboren en la coagulació de la sang quan es produeix la ruptura d’un vas sanguini.
Una cèl·lula mare limfoide es converteix en un limfoblast i, més tard, en un dels tres tipus de limfòcits (glòbuls blancs):
- limfòcits B, que produeixen anticossos per ajudar a combatre les infeccions del cos
- limfòcits T, que ajuden els limfòcits B a produir anticossos per combatre infeccions
- limfòcits citotòxics naturals o limfòcits NK (natural killer), un tipus de cèl·lula immunitària que conté enzimes que poden destruir cèl·lules tumorals o cèl·lules infectades per virus.
En els nens afectats per una leucèmia mieloide aguda, hi ha massa cèl·lules mare que es transformen en mieloblasts.
Quines són les causes de la leucèmia mieloide aguda infantil?
Les causes específiques que originen la majoria dels casos de LMA pediàtrica no es coneixen. Només en un percentatge molt petit de casos (prop del 5 %) les leucèmies agudes en l’edat pediàtrica es desenvolupen en pacients amb una malaltia genètica subjacent amb predisposició a la leucèmia com ara la síndrome de Down o síndromes congènites d’insuficiència medul·lar (l‘Anèmia de Fanconi o la Disqueratosi congènita, entre d’altres).
La leucèmia, com altres tipus de càncer, no és contagiosa. Vegeu apartat Leucèmia, medul·la òssia i cèl·lules sanguínies.
Com es classifica la leucèmia mieloide aguda infantil?
Els dos esquemes més comunament usats per classificar les LMA són: la classificació FAB (franco-americana-britànica), basada en les característiques microscòpiques i en l’expressió de determinades proteïnes en la cèl·lula leucèmica (immunofenotip); i el nou sistema de l’OMS (Organització Mundial de la Salut), que incorpora informació genètica o molecular de la cèl·lula leucèmica i la informació clínica amb interès pronòstic.Al nostre país, en la pràctica clínica habitual, la més estesa és la classificació FAB.
| FAB | Nom | % en pediatria |
|---|---|---|
| M0 | LMA amb poca diferenciació | 2-5 |
| M1 | LMA sense maduració | 10-15 |
| M2 | LMA amb maduració | 25-30 |
| M3 | Leucèmia promielocítica aguda | 5-10 |
| M4 | Leucèmia mielomonocítica aguda | 15-25 |
| M4Eo | M4 + eosinofília en la medul·la òssia | 10 |
| M5 | Leucèmia monoblàstica/monocítica | 15-25 |
| M6 | Leucèmia eritroide | 1-3 |
| M7 | Leucèmia aguda megacarioblàstca | 5-10 |
La classificació de l’OMS, actualitzada el 2022, valora aspectes genètics i moleculars de les cèl·lules leucèmiques. Les alteracions citogenètiques més habituals en les LMA són les translocacions, desplaçament d’un fragment d’un cromosoma a un altre cromosoma (s’indica amb una t). Exemple: t(8; 21), un fragment del cromosoma 8 es desplaça a una zona del cromosoma 21; o dins del mateix cromosoma, t(16; 16). També poden observar-se inversions citogenètiques, que és quan un segment cromosòmic canvia de sentit dins del mateix cromosoma (s’indica com inv).
 Les translocacions o inversions detectades en els estudis citogenètics generen reordenaments dels gens localitzats en les regions cromosòmiques afectades. Així, doncs, es representen amb els noms dels gens implicats, per exemple en el cas de la t(8; 21) generarà un reordenament dels gens RUNX1 i RUNX1T1. Les alteracions citogenètiques han demostrat ser un factor pronòstic molt important i són utilitzades en la majoria de protocols de tractament per determinar-ne la intensitat.En els últims anys s’han anat descrivint mutacions en un o diversos gens de les cèl·lules leucèmiques de la majoria dels pacients. Algunes d’elles han demostrat tenir importància pronòstica i ser rellevants per definir la intensitat del tractament. Així, en la recent classificació de l’OMS s’incorporen les mutacions en NPM1 i CEBPA, associades a un pronòstic més favorable.
Les translocacions o inversions detectades en els estudis citogenètics generen reordenaments dels gens localitzats en les regions cromosòmiques afectades. Així, doncs, es representen amb els noms dels gens implicats, per exemple en el cas de la t(8; 21) generarà un reordenament dels gens RUNX1 i RUNX1T1. Les alteracions citogenètiques han demostrat ser un factor pronòstic molt important i són utilitzades en la majoria de protocols de tractament per determinar-ne la intensitat.En els últims anys s’han anat descrivint mutacions en un o diversos gens de les cèl·lules leucèmiques de la majoria dels pacients. Algunes d’elles han demostrat tenir importància pronòstica i ser rellevants per definir la intensitat del tractament. Així, en la recent classificació de l’OMS s’incorporen les mutacions en NPM1 i CEBPA, associades a un pronòstic més favorable.
| Classificació OMS de la LMA i neoplàsies relacionades |
|---|
| LMA amb alteracions genètiques recurrents |
| LMA amb t(8;21); RUNX1-RUNX1T1 |
| LMA amb inv(16) o t(16;16); CBF-MYH11 |
| LMA amb t(15;17); PML-RARA |
| LMA amb t(9;11); MLLT3-KMT2A |
| LMA amb t(6;9); DEK-NUP214 |
| LMA amb inv(3) o t(3;3);GATA2, MECOM |
| LMA (megacarioblàstica) amb t(1;22); RBM15-MKL1 |
| LMA amb mutació en NPM1 |
| LMA amb mutació bialèlica en CEBPA |
| Entitats provisionals: |
| LMA amb BCR-ABL1 |
| LMA amb mutació en RUNX1 |
| LMA amb canvis en relació amb mielodisplàsia |
| LMA en relació amb tractament |
| LMA no específica (NOS) |
| LMA amb poca diferenciació |
| LMA sense maduració |
| LMA amb maduració |
| Leucèmia mielomonocítica aguda |
| Leucèmia monoblàstica/monocítica |
| Leucèmia eritroide |
| Leucèmia aguda megacarioblàstica |
| Leucèmia aguda basofílica |
| Panmielosi amb mielofibrosi aguda |
| Sarcoma mieloide |
| Proliferació mieloide en relació amb la síndrome de Down |
| Mielopoesi anòmala transitòria |
| Leucèmia mieloide associada a la síndrome de Down |
| Leucèmies agudes de llinatge ambigu |
La majoria dels casos en pediatria es classifiquen dins del grup de LMA amb alteracions genètiques recurrents o LMA no específica.
Quins són els símptomes de la leucèmia mieloide aguda infantil?
La presentació clínica de la leucèmia mieloide aguda és variable i, en general, els símptomes en el diagnòstic es deuen a la infiltració de les cèl·lules leucèmiques de la medul·la òssia i altres òrgans. Encara que pot presentar-se de forma insidiosa, la LMA infantil acostuma a fer-ho de forma aguda, amb una història de menys de tres mesos des de l’inici de la clínica fins al diagnòstic.
En la leucèmia mieloide aguda, la producció de les cèl·lules sanguínies normals es veu alterada pel creixement de les cèl·lules leucèmiques en la mèdul·la òssia. Això pot ocasionar:
- Cansament, debilitat, mareig i pal·lidesa (per anèmia)
- Aparició de morats i petites taques rosades en la pell (petèquies) o altres sagnats (per un recompte de plaquetes baix): hemorràgies nasals, de genives o de qualsevol altres focus.
- Febre i infeccions que no evolucionen bé (degut al mal funcionament dels leucòcits)
En algunes ocasions succeeix el creixement dels ganglis limfàtics, el fetge o la melsa. Pot, així mateix, observar-se simptomatologia específica de la infiltració del sistema nerviós central (mal de cap, vòmits, somnolència, etc.), pell (nòduls disseminats o zones de pell engrossida), mucoses (inflamació de les genives), ocular (visió borrosa, ceguesa ), entre d’altres.

A l’inici de la malaltia, tots aquests símptomes poden ser molt semblants als d’una infecció per un virus. Quan els símptomes continuen més de 2-4 setmanes, en la majoria dels casos, es pot arribar a fer el diagnòstic. Ja que no són símptomes específics o exclusius de la leucèmia, és molt freqüent que s’hagi consultat en diverses ocasions el metge abans que s’arribi al diagnòstic. Generalment, això no influeix en les opcions de curació del nen o nena.
Com es diagnostica la leucèmia mieloide aguda infantil?
A part dels estudis bàsics en sang i medul·la òssia (morfologia, recompte, immunofenotip) que es realitzen en tota leucèmia, els estudis citogenètics (per detectar anomalies cromosòmiques concretes) i estudis moleculars (per detectar alteracions genètiques especifiques) són fonamentals per tipificar i classificar la malaltia. Determinades alteracions genètiques i moleculars es correlacionen amb la sensibilitat al tractament i al risc de recaiguda.
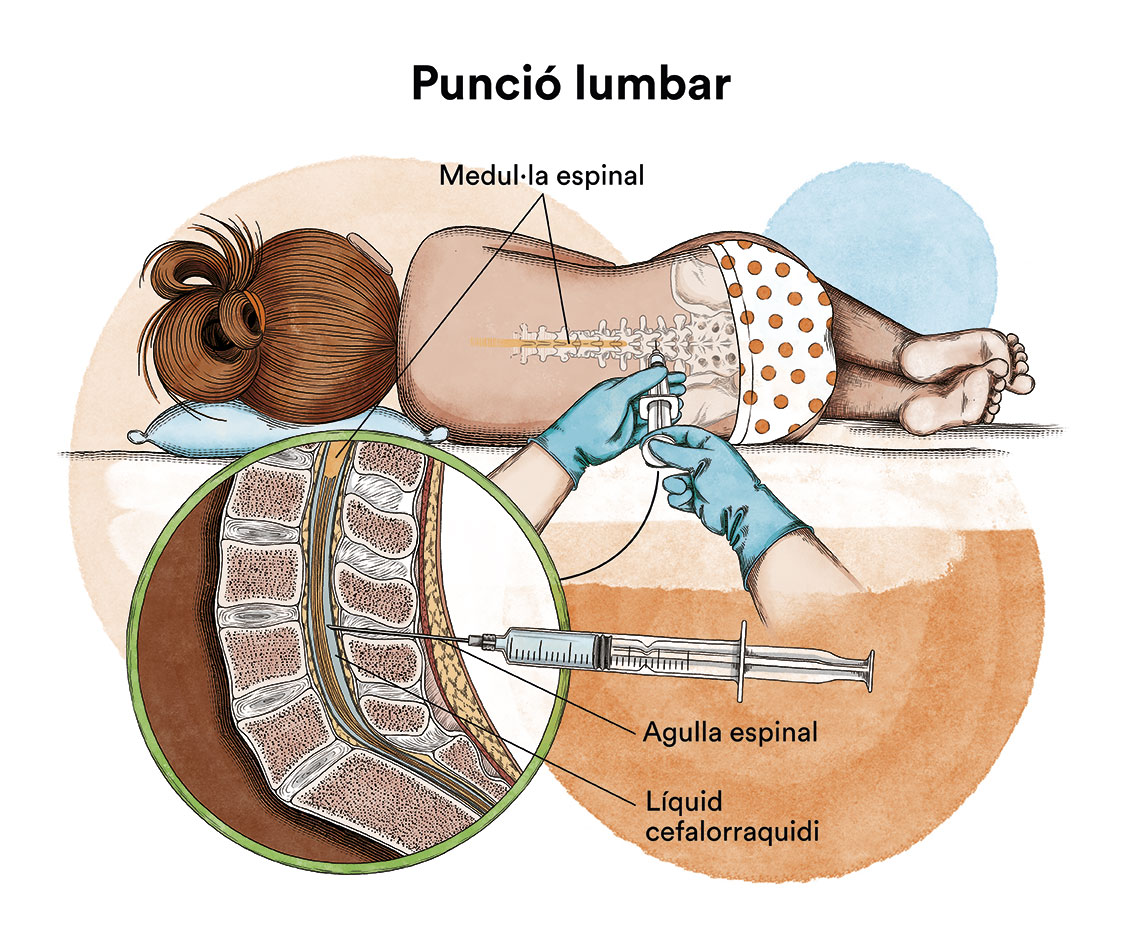
També s’ha d’estudiar si la malaltia s’ha estès al sistema nerviós centralper a la qual cosa s’efectua una punció lumbar amb la finalitat d’analitzar el líquid que envolta aquest sistema (líquid cefalorraquidi).
Quin és el tractament de la leucèmia mieloide aguda en nens?
 L’objectiu del tractament de la leucèmia mieloide aguda és eliminar les cèl·lules leucèmiques per permetre que la medul·la òssia torni a treballar amb normalitat.El pronòstic de les LMA en pediatria ha millorat de forma significativa en les últimes dècades. Aquesta millora ve donada, entre altres, per una millor classificació o estratificació de cada pacient en grups de risc, és a dir, segons el risc individual de recaiguda. Aquesta estratificació permet aplicar estratègies terapèutiques adaptades, per la qual cosa s’intensificarà el tractament en els pacients que tenen uns factors pronòstics d’Alt Risc i es reduirà en aquells de Baix Risc de recaiguda.L’objectiu final del tractament és aconseguir la remissió completa de la malaltia i que sigui profunda (en l’àmbit molecular) i permanent.Diferenciem bàsicament dues fases de tractament: d’inducció i de postremissió o consolidació. La fase de manteniment amb dosis baixes de quimioteràpia utilitzada en els protocols de leucèmia limfoblàstica aguda ha estat abandonada en la majoria de protocols de LMA perquè no aporta una eficàcia addicional, exceptuant en el subgrup de la leucèmia promielocítica aguda (vegeu a continuació). El tractament de la leucèmia mieloide aguda pediàtrica es basa sempre en quimioteràpia intensiva; administració endovenosa de diferents fàrmacs citostàtics (quimioteràpia) al llarg de diversos cicles de tractament. Habitualment, encara que pot variar segons el protocol, es realitzen 1 o 2 cicles d’inducció seguits de 2-3 cicles de consolidació.
L’objectiu del tractament de la leucèmia mieloide aguda és eliminar les cèl·lules leucèmiques per permetre que la medul·la òssia torni a treballar amb normalitat.El pronòstic de les LMA en pediatria ha millorat de forma significativa en les últimes dècades. Aquesta millora ve donada, entre altres, per una millor classificació o estratificació de cada pacient en grups de risc, és a dir, segons el risc individual de recaiguda. Aquesta estratificació permet aplicar estratègies terapèutiques adaptades, per la qual cosa s’intensificarà el tractament en els pacients que tenen uns factors pronòstics d’Alt Risc i es reduirà en aquells de Baix Risc de recaiguda.L’objectiu final del tractament és aconseguir la remissió completa de la malaltia i que sigui profunda (en l’àmbit molecular) i permanent.Diferenciem bàsicament dues fases de tractament: d’inducció i de postremissió o consolidació. La fase de manteniment amb dosis baixes de quimioteràpia utilitzada en els protocols de leucèmia limfoblàstica aguda ha estat abandonada en la majoria de protocols de LMA perquè no aporta una eficàcia addicional, exceptuant en el subgrup de la leucèmia promielocítica aguda (vegeu a continuació). El tractament de la leucèmia mieloide aguda pediàtrica es basa sempre en quimioteràpia intensiva; administració endovenosa de diferents fàrmacs citostàtics (quimioteràpia) al llarg de diversos cicles de tractament. Habitualment, encara que pot variar segons el protocol, es realitzen 1 o 2 cicles d’inducció seguits de 2-3 cicles de consolidació. El tractament d’inducció persegueix l’eliminació de les cèl·lules leucèmiques de la sang i de la majoria de malaltia present a la medul·la òssia a fi de restituir-ne el funcionament normal. A aquest fet se’l denomina aconseguir la remissió completa. Aquesta situació clínica acostuma a aconseguir-se després del primer cicle d’inducció, encara que de vegades pot ser necessari administrar dos cicles d’inducció per aconseguir l’estat de remissió completa. Amb els protocols actuals, més d’un 85 % dels pacients aconseguiran la remissió completa després de la fase d’inducció. Quan la quimioteràpia s’administra per via intravenosa, per evitar punxar repetidament una vena, s’utilitza un dispositiu especial anomenat catèter (contingut en espanyol). El catèter s’introdueix en una vena gran que permet administrar tot tipus de medicaments, així com també extraure sang per a les anàlisis de sang per evitar les repetides puncions al nen.Existeix un tipus de catèter, anomenat port-a-cath (contingut en espanyol), que s’uneix a un reservori rodó de plàstic o metall que queda sota la pell del tòrax. El port-a-cath és molt pràctic en nens perquè al quedar sota la pell no permet que el nen o nena se l’arranqui, és més difícil que s’infecti que altres tipus de catèter i permet que el nen es banyi.
El tractament d’inducció persegueix l’eliminació de les cèl·lules leucèmiques de la sang i de la majoria de malaltia present a la medul·la òssia a fi de restituir-ne el funcionament normal. A aquest fet se’l denomina aconseguir la remissió completa. Aquesta situació clínica acostuma a aconseguir-se després del primer cicle d’inducció, encara que de vegades pot ser necessari administrar dos cicles d’inducció per aconseguir l’estat de remissió completa. Amb els protocols actuals, més d’un 85 % dels pacients aconseguiran la remissió completa després de la fase d’inducció. Quan la quimioteràpia s’administra per via intravenosa, per evitar punxar repetidament una vena, s’utilitza un dispositiu especial anomenat catèter (contingut en espanyol). El catèter s’introdueix en una vena gran que permet administrar tot tipus de medicaments, així com també extraure sang per a les anàlisis de sang per evitar les repetides puncions al nen.Existeix un tipus de catèter, anomenat port-a-cath (contingut en espanyol), que s’uneix a un reservori rodó de plàstic o metall que queda sota la pell del tòrax. El port-a-cath és molt pràctic en nens perquè al quedar sota la pell no permet que el nen o nena se l’arranqui, és més difícil que s’infecti que altres tipus de catèter i permet que el nen es banyi.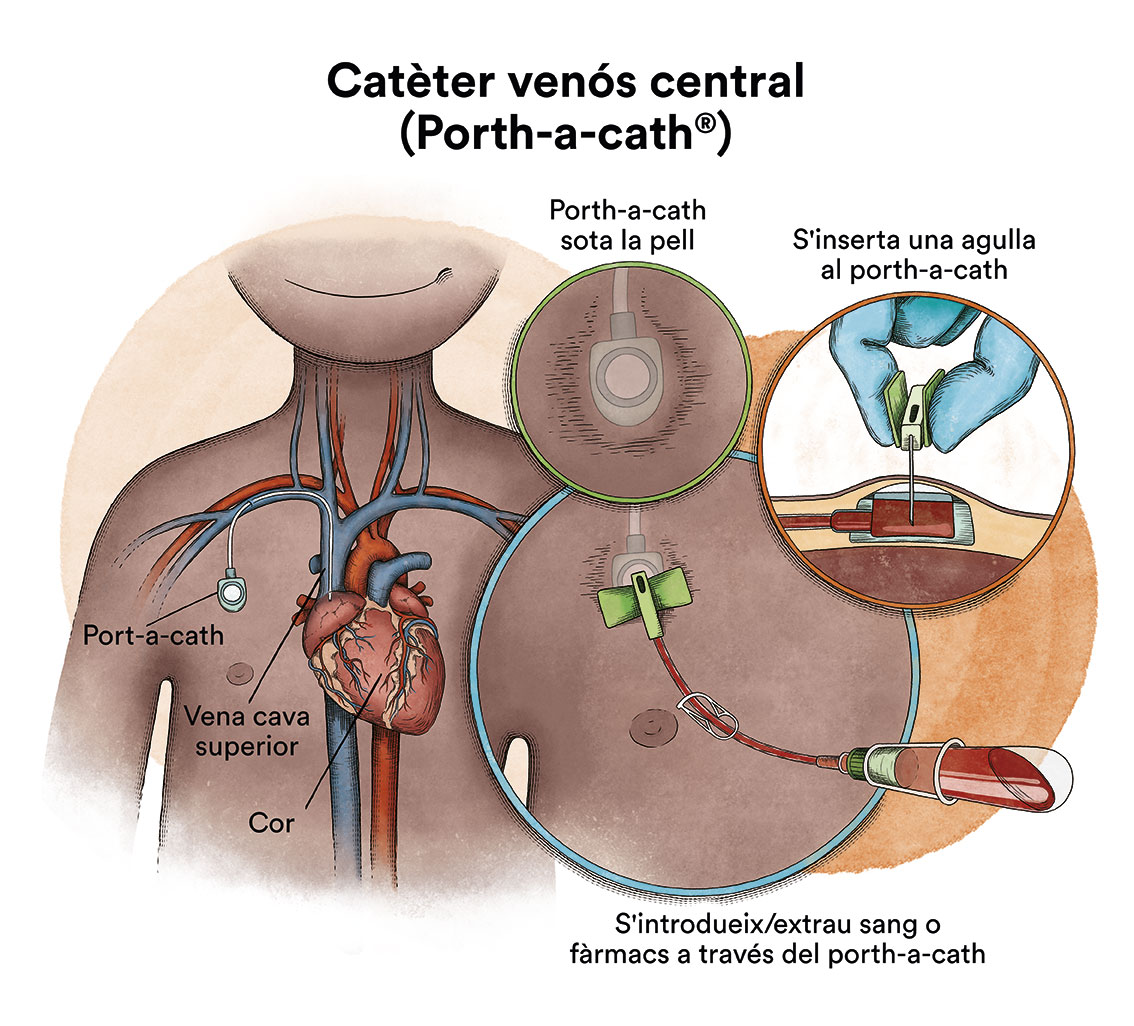 A continuació, s’ha de fer un tractament de postremissió o consolidació que té per objectiu eliminar les cèl·lules leucèmiques residuals (malaltia residual mínima), potencials responsables de la recaiguda de la malaltia.Si la quimioteràpia s’administra per un catèter venós arriba, per la sang, a quasi la totalitat de les cèl·lules del cos. No obstant això, la majoria dels medicaments de la quimioteràpia no arriben bé al líquid cefalorraquidi que banya el cervell i la medul·la espinal. Això fa que hi hagin cèl·lules leucèmiques que poden sobreviure en aquest líquid. Amb la finalitat de prevenir que les cèl·lules leucèmiques que arriben al líquid cefalorraquidi sobrevisquin i siguin la causa d’una futura recaiguda al sistema nerviós, s’ha d’administrar quimioteràpia directament en el líquid cefalorraquidi, mitjançant puncions lumbars (quimioteràpia intratecal). La utilització de radioteràpia cranial ha estat abandonada en la majoria de protocols.En alguns pacients pot estar indicat realitzar un trasplantament de progenitors hematopoètics (TPH) (comunament conegut com a trasplantament de medul·la òssia) com a part del tractament de consolidació. La majoria de protocols actuals han abandonat l’autotrasplantament de medul·la òssia (d’un mateix) per a aquesta patologia i, en cas d’estar indicat, es recomana el TPH procedent d’un donant compatible (TPH al·logènic), ja sigui familiar o no relacionat.
A continuació, s’ha de fer un tractament de postremissió o consolidació que té per objectiu eliminar les cèl·lules leucèmiques residuals (malaltia residual mínima), potencials responsables de la recaiguda de la malaltia.Si la quimioteràpia s’administra per un catèter venós arriba, per la sang, a quasi la totalitat de les cèl·lules del cos. No obstant això, la majoria dels medicaments de la quimioteràpia no arriben bé al líquid cefalorraquidi que banya el cervell i la medul·la espinal. Això fa que hi hagin cèl·lules leucèmiques que poden sobreviure en aquest líquid. Amb la finalitat de prevenir que les cèl·lules leucèmiques que arriben al líquid cefalorraquidi sobrevisquin i siguin la causa d’una futura recaiguda al sistema nerviós, s’ha d’administrar quimioteràpia directament en el líquid cefalorraquidi, mitjançant puncions lumbars (quimioteràpia intratecal). La utilització de radioteràpia cranial ha estat abandonada en la majoria de protocols.En alguns pacients pot estar indicat realitzar un trasplantament de progenitors hematopoètics (TPH) (comunament conegut com a trasplantament de medul·la òssia) com a part del tractament de consolidació. La majoria de protocols actuals han abandonat l’autotrasplantament de medul·la òssia (d’un mateix) per a aquesta patologia i, en cas d’estar indicat, es recomana el TPH procedent d’un donant compatible (TPH al·logènic), ja sigui familiar o no relacionat.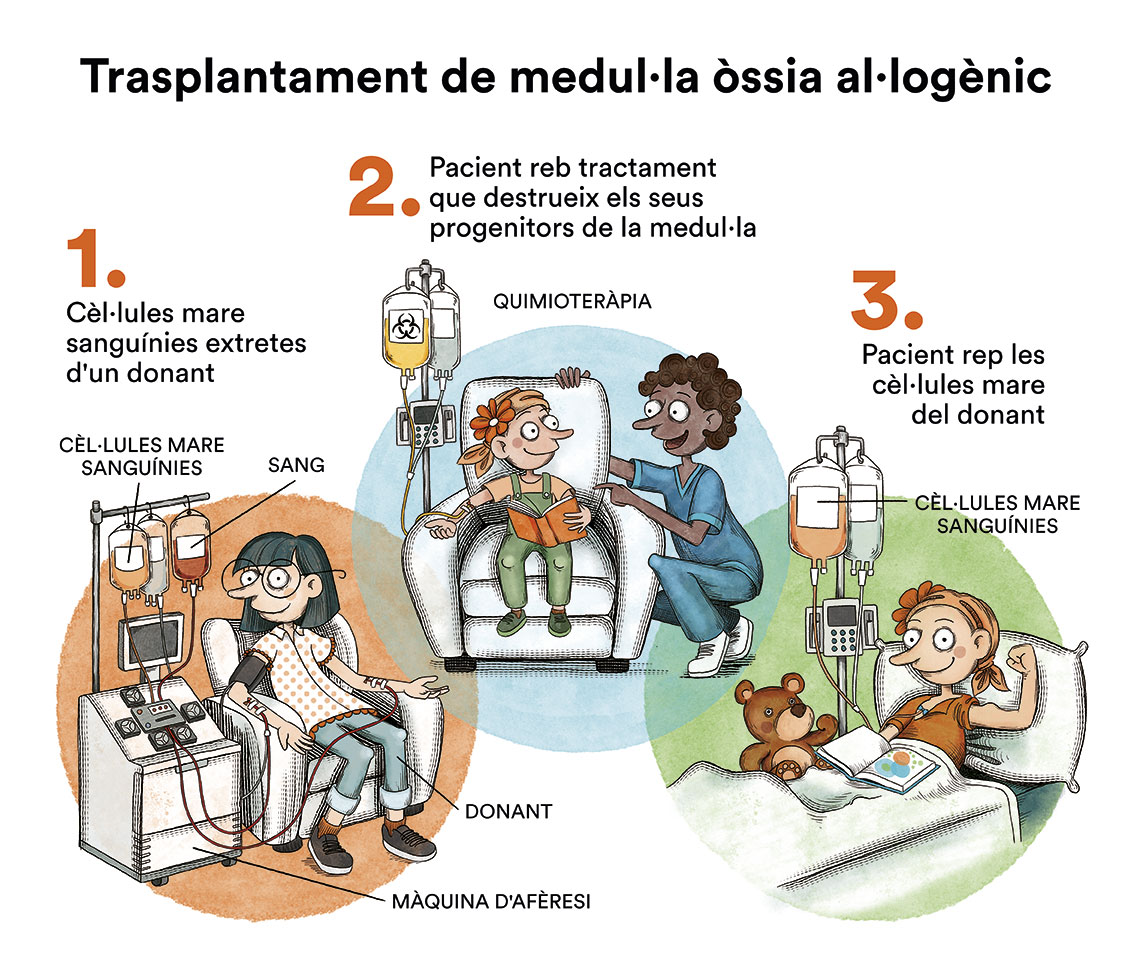 El TPH en LMA pediàtrica és un tema en constant revisió; les indicacions de TPH en pacients amb LMA en primera remissió completa (pacients que no han presentat una recaiguda) són controvertides i no són les mateixes en tots els països. En general, aquest tractament queda reservat per a aquells casos considerats d’alt risc per les característiques biològiques de la malaltia o per una inadequada resposta al tractament quimioteràpic. La majoria de pacients que han recaigut i aconsegueixen un segon estat de remissió completa són candidats a TPH.
El TPH en LMA pediàtrica és un tema en constant revisió; les indicacions de TPH en pacients amb LMA en primera remissió completa (pacients que no han presentat una recaiguda) són controvertides i no són les mateixes en tots els països. En general, aquest tractament queda reservat per a aquells casos considerats d’alt risc per les característiques biològiques de la malaltia o per una inadequada resposta al tractament quimioteràpic. La majoria de pacients que han recaigut i aconsegueixen un segon estat de remissió completa són candidats a TPH.

Quines probabilitats tenen de curar-se els nens amb leucèmia mieloide aguda?
Les probabilitats de curació venen determinades per les característiques del pacient, de la malaltia (alteracions genètiques/moleculars), el tractament que s’administri i per la resposta que es presenti. A diferència dels adults, on les característiques del pacient com ara l’edat avançada o la coexistència d’altres patologies acostumen a ser molt rellevants, no són factors rellevants en l’edat pediàtrica.
La supervivència dels nens i nenes amb LMA ha millorat notablement en els darrers anys, amb taxes de supervivència de prop del 65 %. Aquesta millora ha estat possible gràcies a l’increment en la intensitat del tractament quimioteràpic, una millor classificació dels pacients en grups de risc, la implementació de mesures de suport més eficaces (millors antibiòtics, facilitat per a les transfusions de sang i plaquetes, suport nutricional, infermeria especialitzada ..), així com una notable millora en la selecció de donants per a la realització del TPH.
Nous tractaments contra la leucèmia mieloide aguda infantil
En els últims anys s’està avançant cap a tractaments més personalitzats on es tinguin en compte les característiques de cada individu i de la malaltia (subtipus genètic, molecular, etc). La investigació en aquest camp és molt activa i, per tant, no és d’estranyar que s’hagin desenvolupat nous fàrmacs per a aquestes malalties. La majoria d’aquests fàrmacs encara no formen part dels protocols estàndards de tractament, però molts es troben en fases avançades d’implementació clínica.
Dins de les diferents línies de desenvolupament de nous fàrmacs, cal destacar-ne:
- Nous fàrmacs quimioteràpics: Actuen de manera similar als fàrmacs existents però tenen més eficàcia i/o menys toxicitat. Per exemple, la daunorubicina liposomal permet administrar dosis altes de tractament i, per tant, molt eficaces, però amb una baixa toxicitat sobre el cor, un dels principals inconvenients d’aquest grup de fàrmacs.
- Teràpies dirigides: Són fàrmacs que estan dirigits cap a components específics de les cèl·lules tumorals i tenen un menor impacte sobre les cèl·lules sanes. Dins d’aquest grup destaquem:
- Els anticossos monoclonals, combinen un fàrmac antineoplàstic amb un anticòs que reconeix proteïnes de la cèl·lula tumoral. La identificació d’alteracions citogenètiques-moleculars en la majoria de pacients amb LMA ha permès el desenvolupament de nous fàrmacs que, per diferents mecanismes, actuen sobre aquestes dianes “moleculars” específiques i, per tant, són molt selectius sobre les cèl·lules neoplàstiques.
- La immunoteràpia aprofita les propietats del sistema immunològic propi per actuar sobre les cèl·lules leucèmiques. És una de les àrees de major investigació en els últims anys, però amb escassa aplicació en la LMA.
Subtipus específics
- Leucèmia promielocítica aguda
Una de les leucèmies que més s’ha beneficiat d’una estratègia terapèutica individualitzada és la leucèmia aguda promielocítica. En les últimes dècades, gràcies a la investigació científica, s’ha obtingut una millora substancial en el seu tractament, i ha passat de ser un subtipus de LMA amb molt mal pronòstic a ser una malaltia que respon molt bé al tractament. Aquest tipus de leucèmia es caracteritza perquè té una translocació entre els cromosomes 15 i 17 [t (15:17)], que afecta el receptor de l’àcid retinoic alfa (RARα o RARA) i que li confereix una alta sensibilitat al tractament amb àcid holotransretinoic (ATRA).
- Pacients amb síndrome de Down
Els infants amb síndrome de Down tenen un risc 15 vegades superior a presentar una leucèmia aguda. En el cas de les LMA, l’edat de presentació acostuma a ser per sota dels 5 anys i de forma característica el subtipus que presenten són leucèmia megacarioblàstica aguda (M7, segons classificació FAB) o leucèmia eritroide aguda (M6, segons classificació FAB).
Aquest grup de pacients presenta una elevada sensibilitat als tractaments quimioteràpics i això ha possibilitat taxes de curació elevades. Una de les principals dificultats per aconseguir la curació es deu a l’elevada toxicitat davant alguns fàrmacs quimioteràpics i l’elevat risc d’infecció. És per això que diferents grups han aconseguit augmentar la supervivència amb protocols de tractaments adaptats.
Fins a un 10 % dels nens i nenes amb síndrome de Down presenten una proliferació transitòria de cèl·lules leucèmiques durant els primers mesos de vida. Aquestes cèl·lules són morfològicament indistingibles d’una LMA. Aquest fenomen es coneix com a síndrome mieloproliferatiu transitori o mielopoesi anòmala transitòria. Habitualment presenten un curs benigne i solen involucionar espontàniament durant els tres primers mesos de vida, encara que alguns pacients poden requerir tractaments amb dosis baixes de citostàtics. És important el seguiment posterior, ja que un 20 % d’aquests nens desenvoluparan una LMA durant els tres primers anys de vida.
Seguiment
Després de completar el tractament, el nen seguirà controls periòdics pel seu metge hematòleg i per altres especialistes en cas que sigui necessari. Els controls es realitzen per avaluar una possible recaiguda i per fer un seguiment i un tractament de les possibles complicacions a llarg termini. Aquests controls es van espaiant progressivament fins fer-se una vegada a l’any. És recomanable fer un seguiment com a mínim anual a llarg termini per poder detectar aviat i poder tractar, si apareixen, les seqüeles del tractament o de la leucèmia.
Recomanacions i altres aspectes pràctics
A continuació, farem unes recomanacions de caràcter general i que responen a algunes de les preguntes més freqüents que realitzen els pares dels nens amb leucèmia:
- Se li cauran els cabells? Quan? Els hem de tallar?
Amb la quimioteràpia que rebrà per tractar la leucèmia, els cabells li cauran. Generalment, això succeeix a les 2-3 setmanes de l’inici de la quimioteràpia. Si el nen o la nena té els cabells llargs, és més adequat tallar-los curts abans que comencin a caure. No cal, ni és convenient des del punt de vista psicològic, tallar-los durant els primers dies de l’ingrés. Tampoc cal explicar-li aquest fet en un primer moment. Sí convé, però, abordar aquest tema amb el nen abans que comencin a caure. Els cabells tornen a créixer al cap de 2-4 setmanes d’haver iniciat la fase de tractament de manteniment, en la qual la quimioteràpia és de menys intensitat.
- Higiene
Degut a que el nen té disminuïdes les seves defenses davant les infeccions (per la pròpia malaltia i també pel tractament administrat), és convenient mantenir una higiene corporal adequada de l’infant, de l’habitació de l’hospital i del domicili familiar, així com de les seves joguines .
És recomanable evitar aquelles joguines que emmagatzemen molta pols i les caixes de cartró. Tampoc s’ha d’emmagatzemar menjar fora de la nevera. Les plantes estan prohibides a l’habitació, ja que a la terra hi ha espores de fongs.
L’ordre facilita la neteja per part del personal de la neteja de l’hospital.
- Visites
És convenient reduir el nombre de visites a l’habitació del nen, ja que poden ser portadores d’infeccions. És recomanable que no hi hagi més de 2 acompanyants a l’habitació i que es rentin les mans abans d’entrar. Si algun dels visitants té algun procés infecciós (refredat, conjuntivitis …) és preferible que no vingui.
En cas que aquest sigui el pare, la mare o una altra persona que tingui cura del nen i que no es pugui prescindir de la seva atenció, convindria que es posessin una mascareta i es rentessin les mans abans d’entrar en contacte amb el nen.
- Alimentació
El nen que rep tractament amb quimioteràpia intensiva ha de rebre una alimentació variada. És convenient, quan la xifra de leucòcits és baixa, evitar els aliments crus que no es puguin pelar (per exemple: enciam, maduixes, tomàquet cru).
De vegades la quimioteràpia pot treure la gana, o fins i tot provocar nàusees. Durant els dies que estigui rebent la quimioteràpia, no convé forçar el nen perquè mengi, perquè pot ser contraproduent.
D’altra banda, els corticoides (prednisona i dexametasona) que rebrà en algunes fases del tractament poden augmentar molt la gana, fins i tot amb ansietat. Si bé se’ls pot permetre menjar alguna cosa més que els menjars que se serveixen a l’hospital, no se’ls ha de deixar menjar sense límit, ja que sovint no ho toleren bé i pot ocasionar mal de panxa.
Enllaços d'interès sobre temes mèdics relacionats amb la leucèmia mieloide aguda en nens
- Tractament de la leucèmia mieloide aguda en nens. National Cancer Institute.
- Informació sobre la leucèmia promielocítica aguda. Leukemia and Lymphoma Society
- La leucèmia mieloide aguda en nens i adolescents. Leukemia and Lymphoma Society
Enllaços d'interès sobre altres temes relacionats amb la leucèmia mieloide aguda en nens
MATERIALS LEUCÈMIA INFANTIL
- Els nadons també tenen leucèmia. Fundació Josep Carreras contra la leucèmia. (contingut en espanyol)
- Leucèmia infantil. Els petits imparables. Fundació Josep Carreras contra la leucèmia. (contingut en espanyol)
- Joc retallable Medulín. Fundació Josep Carreras contra la leucèmia. (contingut en espanyol)
La Fundació Josep Carreras disposa d’un conte “El nadó forçut” dirigit a nens o germans que pateixen leucèmia. Està especialment dirigit a nens fins als 6 anys. Si el vols demanar, pots enviar-nos un correu electrònic a imparables@fcarreras.es.
TRASPLANTAMENT DE MEDUL·LA ÒSSIA
- Guia del Trasplantament de Medul·la Òssia. Fundació Josep Carreras (contingut en espanyol)
- Què és l’HLA i com funciona? Fundació Josep Carreras (contingut en espanyol)
- La Malaltia Empelt contra Receptor. Fundació Josep Carreras (contingut en espanyol)
- Història del Trasplantament de Medul·la Òssia. Fundació Josep Carreras (contingut en espanyol)
- Com es realitza la cerca d’un donant compatible anònim? Fundació Josep Carreras (contingut en espanyol)
MANUALS DE SUPORT
- Com enfrontar-se a la leucèmia i el limfoma en nens? Leukemia & Lymphoma Society.
- VIURE APRENENT. Protocol d’actuació per a alumnes amb càncer AFANION.
- Guia de suport per a pares de nens oncològics ASION.
- Guia per a joves i adolescents amb càncer ASION.
- Alumnat amb càncer. Guia per a docents ASION.
- La importància del comportament dels pares quan un nen té càncer ASION.
- El meu fill té càncer. Què faig? FARO.
ALIMENTACIÓ
- Com mantenir una alimentació saludable durant el tractament? Fundació Josep Carreras (contingut en espanyol)
- “Buon profit”. Consells dietètics durant el tractament AFANION.
- “Les receptes màgiques de Jabel”. Isabel Rojas Murcia, Carolina Mangas Gallardo.
ALTRES
- Informació sobre els efectes a llarg termini i tardans del tractament per a la leucèmia o el limfoma en els nens Leukemia & Lymphoma Society.
- El meu germà té càncer Fundació Josep Carreras contra la leucèmia. (contingut en espanyol)
- L’escola en un hospital Fundació Josep Carreras contra la leucèmia. (contingut en espanyol)
- Educant il·lusions. Guia per a la intervenció psicoeducativa en nens i adolescents amb càncer FARO.
- El càncer en l’adolescència Fundació Josep Carreras contra la leucèmia. (contingut en espanyol)
- Documental “La leucèmia i els adolescents” Fundació Josep Carreras contra la leucèmia. (contingut en espanyol)
- Documental “Els nadons també tenen leucèmia” Fundació Josep Carreras contra la leucèmia. (contingut en espanyol)
- 7 formes de posar-se un mocador Fundació Josep Carreras contra la leucèmia. (contingut en espanyol)
- Conte “La princesa Luzie i els cavallers de la quimio’ ASPANAFOA.
- Conte “Anem a quimioteràpia’.
- Conte “Anem a radioteràpia’.
- Conte “Gasparín Súper Quimio” Federación Española de Padres de Niños con Cáncer.
- Vídeo “Charlie Brown i la leucèmia”.
- Conte “El Toby i la màquina voladora”.
- Conte “La fada de les estrelles” AECC.
- Conte “La Lina, la petita oreneta” Osakidetza.
Enllaços d'interès: entitats locals (recursos i serveis)
Totes aquestes organitzacions són externes a la Fundació Josep Carreras.
ANDALUSIA
ARAGÓ
ASTÚRIES
CASTELLA-LA MANXA
CASTELLA I LLEÓ
CATALUNYA
COMUNITAT VALENCIANA
EXTREMADURA
GALÍCIA
ILLES BALEARS
ILLES CANÀRIES
LA RIOJA
MADRID
- AAA (asociación de adolescentes y Adultos Jóvenes con Cáncer)
- ASION
- FUNDACIÓN CAICO
- FUNDACIÓN ALADINA
- FUNDACIÓN UNOENTRECIENMIL
MÚRCIA
NAVARRA
PAÍS BASC
Suport i ajuda
Et convidem també a seguir-nos a les nostres xarxes socials principals (Facebook, Twitter i Instagram) on sovint compartim testimonis de superació.Si resideixes a l’Estat espanyol, també pots posar-te en contacte amb nosaltres enviant-nos un correu electrònic a imparables@fcarreras.es perquè t’ajudem a posar-te en contacte amb altres persones que han superat aquesta malaltia.