Información avalada por ![]()

¿Qué es la leucemia, la médula ósea y cuáles son los tipos de células sanguíneas?
La leucemia es un tipo de cáncer de las células de la sangre que se origina en la médula ósea. Ver apartado Leucemia, médula ósea y células sanguíneas.
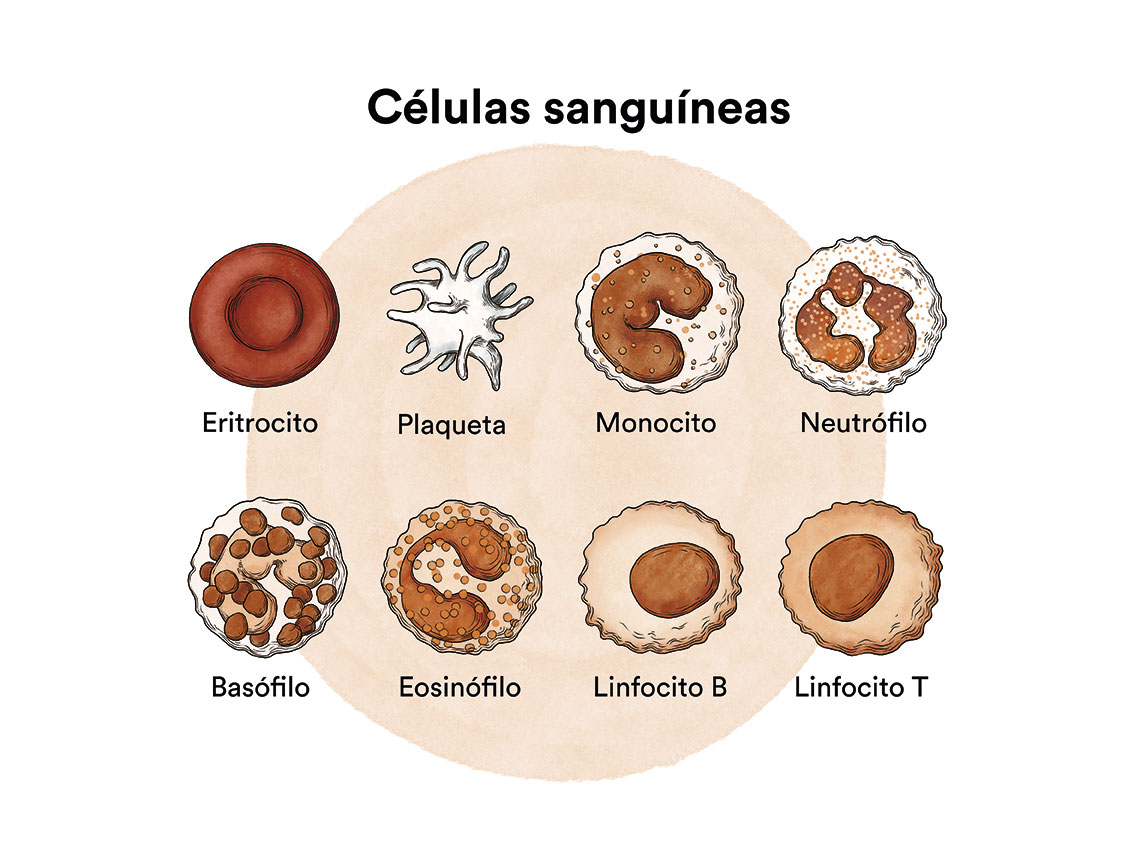
Como hemos visto en ‘Leucemia, médula ósea y células sanguíneas’, la médula ósea elabora las células madre sanguíneas (células inmaduras) que, con el tiempo, se transformarán en células sanguíneas maduras. Una célula madre sanguínea se convierte en una célula madre mieloide o en una célula madre linfoide.
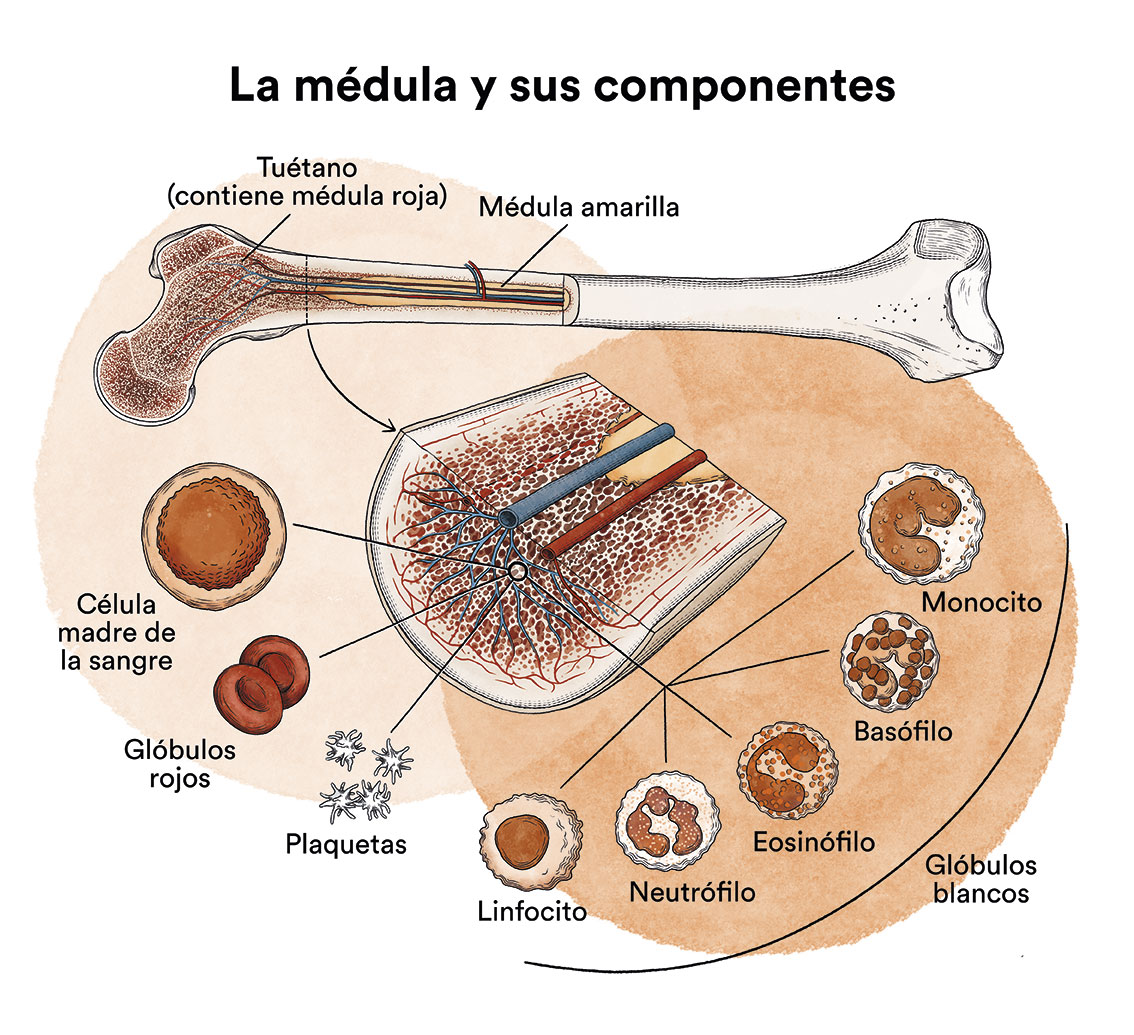
Una célula madre mieloide se convierte en uno de los tres tipos de glóbulos sanguíneos maduros:
- Glóbulos rojos, que transportan el oxígeno a otros tijos y órganos del cuerpo
- Granulocitos, glóbulos blancos que ayudan a combatir infecciones y otras enfermedades
- Plaquetas, que colaboran en la coagulación de la sangre evitando sangrados.
Las leucemias agudas son un grupo de enfermedades neoplásicas caracterizadas por la transformación maligna y producción incontrolada de células hematopoyéticas inmaduras. Existen dos tipos principales en función de la línea celular afectada: la leucemia linfoblástica aguda (LAL) y la leucemia mieloblástica o mieloide aguda (LMA).
¿Qué es la leucemia mieloide aguda en adultos y a quién afecta?
La leucemia mieloide aguda (también conocida como leucemia mieloblástica aguda, leucemia mielógena aguda, leucemia granulocítica aguda o LMA) es el tipo más común de leucemia aguda en adultos, si bien en ocasiones puede diagnosticarse en niños. La mediana de edad de aparición son 64 años y, la mayoría de los pacientes, se sitúan en la franja de los 60-75 años. Este tipo de leucemia representa el 40% de todas las leucemias en el mundo occidental. Su incidencia en nuestro país se estima en 15 nuevos casos por millón de habitantes y año.
 En ocasiones, la leucemia mieloide aguda es la etapa final de otras enfermedades como los síndromes mielodisplásicos o los síndromes mieloproliferativos crónicos. De igual forma, la leucemia mieloide aguda puede aparecer años después de haber recibido quimioterapia y/o radioterapia para el tratamiento de otra neoplasia; estas leucemias mieloides agudas clásicamente se han denominado leucemias secundarias.
En ocasiones, la leucemia mieloide aguda es la etapa final de otras enfermedades como los síndromes mielodisplásicos o los síndromes mieloproliferativos crónicos. De igual forma, la leucemia mieloide aguda puede aparecer años después de haber recibido quimioterapia y/o radioterapia para el tratamiento de otra neoplasia; estas leucemias mieloides agudas clásicamente se han denominado leucemias secundarias.
Otra particularidad es su incidencia es más elevada entre personas con determinadas alteraciones cromosómicas como el síndrome de Down o la Anemia de Fanconi.
¿Cuáles son las causas de la leucemia mieloide aguda en adultos?
Las causas específicas que originan la mayoría de los casos de leucemia mieloide aguda no se conocen. No obstante, existen algunos factores de riesgo que se asocian con una probabilidad más alta de desarrollar una leucemia mieloide aguda. Un factor de riesgo es todo aquello que aumenta la probabilidad de que una persona pueda desarrollar cáncer.
Los factores de riesgo asociados a la leucemia mieloide aguda son, según la American Society of Clinical Oncology:
- La edad. La leucemia mieloide aguda es más frecuente en los adultos mayores, pero se presenta en todas las edades.
- Trastornos genéticos. La leucemia mieloide aguda ocurre más frecuentemente en personas con los siguientes trastornos heredados:
- Síndrome de Down
- Ataxia-telangiectasia
- Síndrome de Li-Fraumeni (en inglés)
- Síndrome de Klinefelter
- Anemia de Fanconi
- Síndrome de Wiskott-Aldrich
- Síndrome de Bloom
- Síndrome de trastorno plaquetario familiar
- Mutaciones de la línea germinal que están presentes al momento del nacimiento; las más frecuentes son cambios en los genes GATA2, ETV6, CEBPA y RUNX1.
- Dosis altas de radiación. Las personas que han estado expuestas a niveles altos de radiación pueden ser más propensas a desarrollar una leucemia mieloide aguda. Esto incluye a personas que han recibido radioterapia para otro cáncer o supervivientes a largo plazo de bombas atómicas o accidentes de fugas de radioactividad. No se ha demostrado que los campos electromagnéticos generados por líneas de energía eléctrica de alto voltaje causen leucemia mieloide aguda. El uso de teléfonos celulares no es un factor de riesgo de leucemia mieloide aguda conocido.
- Quimioterapia. Las personas que han recibido quimioterapia para otro cáncer pueden desarrollar una leucemia mieloide aguda relacionada con la terapia anterior.
- Sustancias químicas. El contacto prolongado con productos que contienen benceno (que se encuentra en el petróleo, el humo de cigarrillo y algunos lugares de trabajo industriales) aumenta el riesgo de leucemia mieloide aguda. Sin embargo, no se ha demostrado que la exposición a disolventes industriales y a tintes capilares aumente el riesgo de una persona de desarrollar leucemia.
- Otros trastornos de la médula ósea. Es posible que las personas que tengan otros trastornos de la médula ósea, incluidos trastornos mieloproliferativos, desarrollen leucemia mieloide aguda con el tiempo. “Mielo” significa de la médula ósea y “proliferativo” significa multiplicarse en abundancia. Estas afecciones incluyen: Leucemia Mieloide Crónica, Policitemia vera, Mielofibrosis y Trombocitosis esencial. También existe un mayor riesgo de progresión a una LMA en personas con Síndromes mielodisplásicos o Anemia aplásica.
La leucemia, como otros tipos de cáncer, no es contagiosa. Ver apartado Leucemia, médula ósea y células sanguíneas.
¿Cómo se clasifica la leucemia mieloide aguda?
En los años ’70 un grupo de expertos franceses, estadounidenses y británicos definió la clasificación FAB, diferenciando las LAM en subtipos, del M0 al M7, según el tipo de célula del cual la leucemia se desarrolla y cuán maduras están las células. Esto se basó principalmente en la apariencia de las células leucémicas en el microscopio después de una tinción de rutina y en la expresión de determinadas proteínas en la célula leucémica (inmunofenotipo).
| Tipo FAB | Definición | Frecuencia |
|---|---|---|
| LMA 0 | Leucemia mieloide aguda sin diferenciación localizada | 2-5% |
| LMA 1 | Leucemia mieloide aguda sin maduración | 15-20% |
| LMA 2 | Leucemia mieloide aguda con maduración | 25-30% |
| LMA 3 | Leucemia promielocítica aguda (con translocación t15;17) | 10-15% |
| LMA 4 | Leucemia mielomonocítica aguda (LMMA) | 15-30% |
| LMA 5 | Leucemia monocítica aguda (LMoA) | 10-15% |
| LMA 6 | Eritroleucemia | 3-4% |
| LMA 7 | Leucemia megacariocítica aguda | 1% |
Sin embargo, actualmente existen dos nuevas clasificaciones recientemente actualizadas (WHO 2022 e ICC 2022) que se centran en las alteraciones citogenéticas y moleculares de la LAM, las cuales tienen mucha relación con el pronóstico, y pueden ser susceptibles de algún tratamiento dirigido con fármacos diana.
Las alteraciones citogenéticas más habituales en las LMA son las translocaciones; desplazamiento de un fragmento de un cromosoma a otro cromosoma (se indica como t). Ejemplo: t(8;21), un fragmento del cromosoma 8 se desplaza a una zona del cromosoma 21; o dentro del mismo cromosoma, t(16;16). También pueden observarse inversiones citogenéticas que es cuando un segmento cromosómico cambia de sentido dentro del propio cromosoma (se indica como inv).
Las translocaciones o inversiones detectadas en los estudios citogenéticos generan reordenamientos de los genes localizados en las regiones cromosómicas afectadas. Éstos se representan por los nombres de los genes implicados, así en el caso de la t(8;21) generará un reordenamiento de los genes RUNX1 y RUNX1T1. Las alteraciones citogenéticas han demostrado ser un factor pronóstico muy importante y son utilizadas en la mayoría de los protocolos de tratamiento para determinar su intensidad.
En los últimos años se han ido describiendo mutaciones en uno o varios genes de las células leucémicas de la mayoría de los pacientes. Algunas de ellas han demostrado tener importancia pronóstica y ser relevantes para definir la intensidad del tratamiento
Por ello, hoy en día, se valoran tanto las alteraciones cromosómicas como las moleculares para establecer los protocolos de tratamiento.
Conocer el subtipo de leucemia mieloide aguda es muy importante, ya que afecta tanto al pronóstico de un paciente como a la elección del mejor tratamiento. Por ejemplo, el subtipo de leucemia promielocítica aguda (LMA3) se trata a menudo con medicamentos que son diferentes a los utilizados para otros subtipos de leucemia mieloide aguda.
Según las anomalías cromosómicas (citogenéticas) puedes establecerse algunos subtipos de leucemia mieloide aguda con pronóstico más o menos favorable. Estos factores pronósticos ayudan a los hematólogos a determinar el riesgo de que la leucemia de una persona regrese después del tratamiento, y por lo tanto si debe recibir un tratamiento más o menos intensivo. Algunos de estos criterios son:
- Anomalías favorables:
- Translocación entre los cromosomas 8 y 21 (visto con más frecuencia en pacientes con LMA2)
- Translocación o inversión del cromosoma 16
- Translocación entre los cromosomas 15 y 17 (visto con más frecuencia en pacientes con LMA3 – leucemia promielocítica aguda)
- Anomalías desfavorables:
- Deleción (pérdida) de parte del cromosoma 5 o 7
- Translocación o inversión del cromosoma 3
- Translocación entre los cromosomas 6 y 9
- Translocación entre los cromosomas 9 y 22
- Anomalías del cromosoma 11 (en el lugar q23)
- Pérdida de un cromosoma, por lo que la célula tiene solo una copia en lugar de las dos normales (monosomía)
- Cambios complejos (que implican a 3 o más cromosomas)
Los cambios moleculares en el momento del diagnóstico también son muy importantes para comprender el pronóstico y las opciones de tratamiento junto con los cambios cromosómicos. Los cambios genéticos moleculares más frecuentes relacionados con el pronóstico para las personas con LMA incluyen: NPM1, CEBP alfa, FLT3, RUNX1, ASXL1, TP53, IDH1 e IDH2.

¿Cuáles son los síntomas de la leucemia mieloide aguda en adultos?
El intervalo entre la aparición de los primeros síntomas y el diagnóstico es habitualmente inferior a 3 meses debido al carácter agudo de la enfermedad. Los síntomas de los pacientes con leucemia mieloide aguda son consecuencia de la anemia producida por el déficit de glóbulos rojos (sensación de cansancio, debilidad, mareos, palidez); del déficit de plaquetas (hematomas, hemorragias de encías, nasales o de cualquier otro foco); y del déficit de granulocitos (fiebre e infecciones).
Ver leucemia, médula ósea y células sanguíneas.
En ocasiones puede observarse el crecimiento de los ganglios linfáticos, el hígado o el bazo. Puede asimismo observarse sintomatología específica de la infiltración del sistema nervioso central (dolor de cabeza, vómitos, somnolencia, etc.), piel (nódulos diseminados o zonas de piel engrosada), mucosas (inflamación de las encías), ocular (visión borrosa, ceguera), entre otras.
¿Cómo se diagnostica la leucemia mieloide aguda en adultos?
Además de los estudios básicos en sangre y medula ósea (morfología, recuento, inmunofenotipo) a realizar en toda leucemia, los estudios citogenéticos (para detectar anomalías cromosómicas concretas) y estudios moleculares (para detectar alteraciones genéticas especificas) son fundamentales para tipificar y clasificar la enfermedad. Determinadas alteraciones citogenéticas y moleculares se correlacionan con la sensibilidad al tratamiento y el riesgo de recaída.
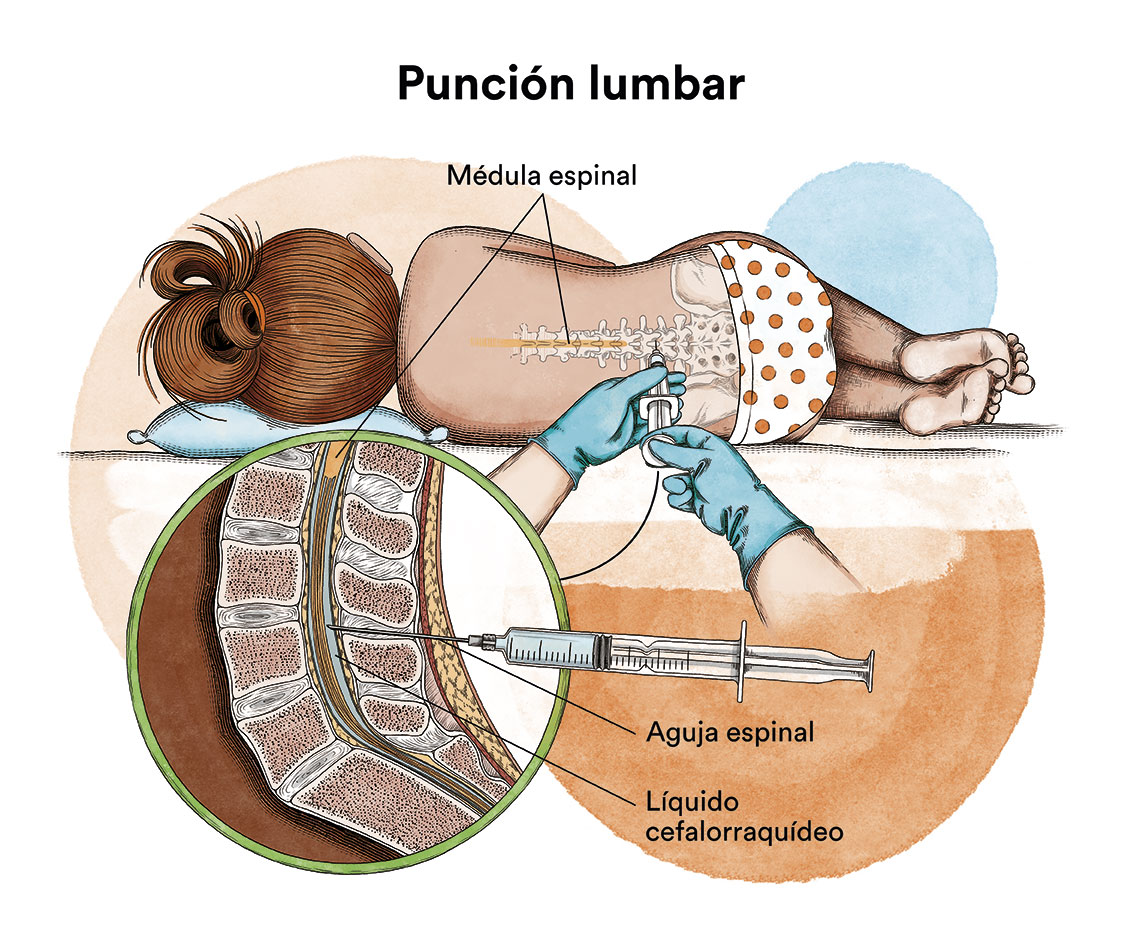
En función de los síntomas y del subtipo de leucemia, en ocasiones debe estudiarse si la enfermedad se ha extendido al sistema nervioso central efectuando, para ello, una punción lumbar con el fin de analizar el líquido que envuelve dicho sistema (líquido cefalorraquídeo), y si estuviera afectado, habría que administrar quimioterapia local (intratecal).
¿Cuál es el tratamiento de la leucemia mieloide aguda en adultos?
El tratamiento de la leucemia mieloide aguda se determinará de forma individualizada teniendo en cuenta el subtipo de la enfermedad, la edad, el estado general del paciente y, posteriormente, la respuesta al tratamiento inicial.
El objetivo principal de cualquier tratamiento en las leucemias u otras hemopatías malignas es conseguir la remisión completa de la enfermedad a nivel molecular.
El tratamiento quimioterápico más utilizado hasta la fecha en pacientes menores de 70 años incluye 2 fases inducción a la remisión y de post-remisión o consolidación. La fase de mantenimiento con dosis bajas de quimioterapia tan utilizada durante periodos muy largos en la leucemia linfoblástica aguda (LLA), no se ha mostrado eficaz en la leucemia mieloide aguda.
Por lo general, el tratamiento de la leucemia mieloide aguda debe comenzarse tan pronto como sea posible después del diagnóstico, si bien, en función de la agresividad de presentación, podemos esperar unos días para tener la información completa sobre las características principales de la enfermedad y así dirigir mejor el tratamiento, siempre que el estado general del paciente sea bueno y haciendo un seguimiento clínico estrecho.
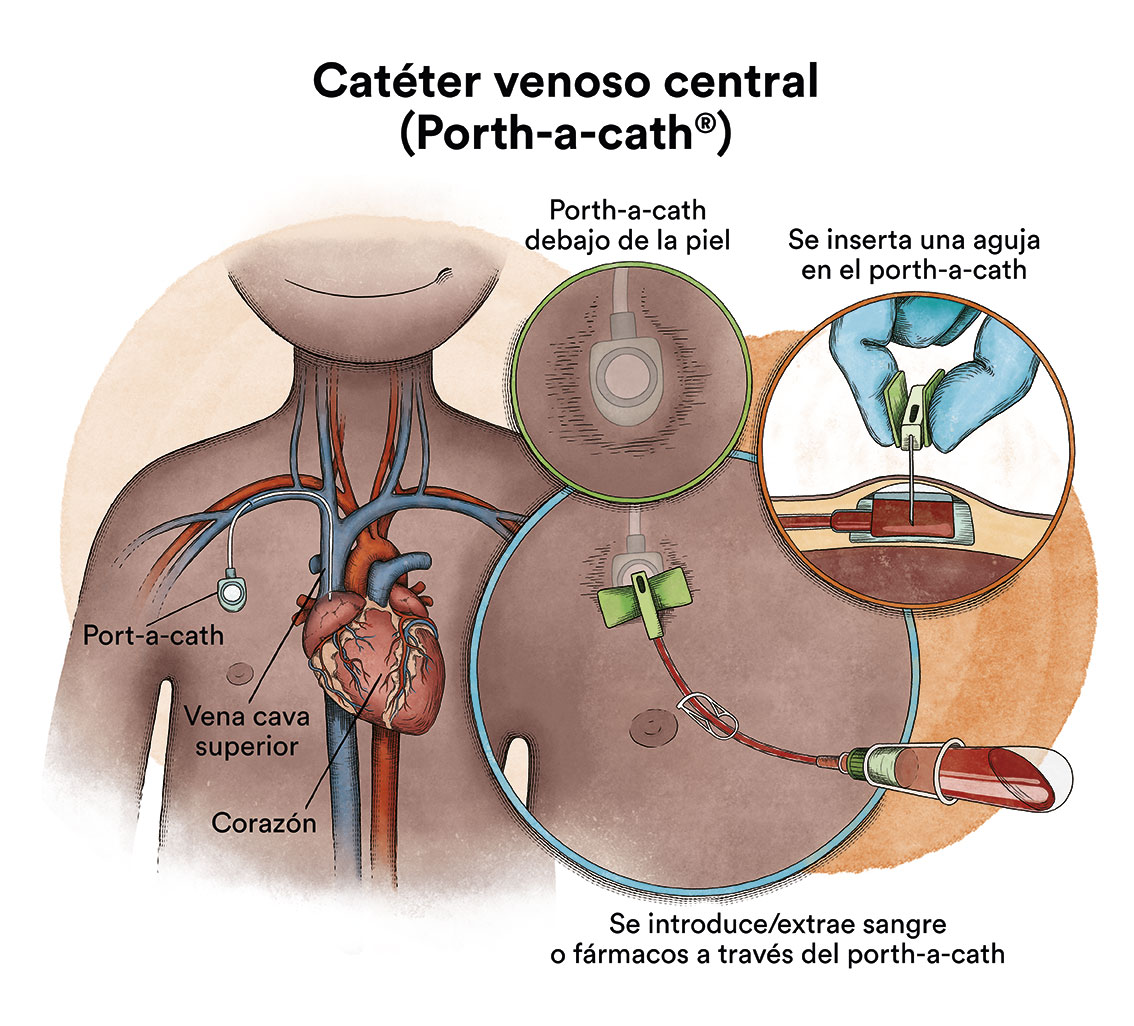
Debido a la administración de quimioterapia y/o de transfusiones sanguíneas, en general, uno de los primeros pasos tras el diagnóstico será la colocación de un catéter venoso central para facilitar el tratamiento, debido a que las venas periféricas (de los brazos) no son suficientemente resistentes para soportar una vía por donde administrar el tratamiento y soporte general (sueros, transfusiones, quimioterapia, etc).
1. Fase de inducción a la remisión (excepto en la leucemia promielocítica aguda. Ver más adelante)
El equipo de hematología considerará el tratamiento a emplear en función de la edad y el estado general del paciente y la presencia de otras enfermedades (llamadas comorbilidades), además de otros aspectos como las alteraciones genéticas o cromosómicas concretas de la leucemia.
El tratamiento estándar de inducción en pacientes sin comorbilidades relevantes y hasta los 70 años, se basa habitualmente en una quimioterapia intensiva que incluye diversos agentes antineoplásicos por vía intravenosa, con el objetivo de lograr que desaparezcan las células leucémicas de la sangre y la médula ósea (remisión completa citológica), permitiendo la producción normal de las otras células sanguíneas. Se considera que un paciente ha alcanzado la remisión completa citológica cuando la cifra de blastos en la médula ósea es inferior al 5 %. Esta situación clínica suele alcanzarse en el 70-80% del total de pacientes tras el primer ciclo de tratamiento, si bien en ocasiones puede ser necesario administrar dos ciclos de inducción para alcanzar la remisión o incluso cambiar de estrategia terapéutica.
 Los dos agentes quimioterápicos empleados son: la citarabina (ara-C) y un medicamento de la familia de las antraciclinas, como daunorrubicina (daunomycin) o idarrubicina. A menudo, esta pauta se llama régimen de 7 + 3, ya que consiste en recibir la citarabina continuamente durante 7 días, junto con infusiones cortas de antraciclina en cada uno de los 3 primeros días.
Los dos agentes quimioterápicos empleados son: la citarabina (ara-C) y un medicamento de la familia de las antraciclinas, como daunorrubicina (daunomycin) o idarrubicina. A menudo, esta pauta se llama régimen de 7 + 3, ya que consiste en recibir la citarabina continuamente durante 7 días, junto con infusiones cortas de antraciclina en cada uno de los 3 primeros días.
En algunas situaciones donde existe una molécula diana un tercer medicamento podría añadirse también para tratar de mejorar las probabilidades de remisión:
- En caso de estar presente la mutación en el gen FLT3, se podría administrar el medicamento de terapia dirigida midostaurin (Rydapt®) junto con quimioterapia. Este medicamento se administra dos veces al día en forma de pastilla. Se están desarrollando también nuevos tratamientos, con esta misma molécula diana, en forma de ensayo clínico hasta que obtengan su aprobación, si procede, y que generalmente se administran por vía oral, y junto a la quimioterapia intravenosa.
- Para pacientes cuyas células leucémicas expresan la proteína CD33, el medicamento de terapia dirigida ozogamicina gemtuzumab (Mylotarg®) se podría agregar a la quimioterapia en pacientes de riesgo favorable o intermedio.
- Cabe remarcar que, en algunos perfiles concretos de pacientes jóvenes, el tratamiento de elección puede no ser el 7+3 sino VYXEOS (una adaptación del 7+3 aprobado en las leucemias secundarias en relación a quimioterapia previa o bien con antecedente de mielodisplasia leucemia mieloide aguda relacionada con el tratamiento (LMA-t) o LMA con cambios relacionados con mielodisplasia) o la combinación de venetoclax con azacitidina.
En los casos en los que la leucemia se ha propagado al sistema nervioso, también se puede administrar quimioterapia en el líquido cefalorraquídeo mediante punciones lumbares (quimioterapia intratecal).
Cabe remarcar que la primera fase de tratamiento suele requerir de un ingreso hospitalario prolongado de aproximadamente 4 o 5 semanas.
 2. Fase de tratamiento de post-remisión o consolidación
2. Fase de tratamiento de post-remisión o consolidación
Esta fase tiene por finalidad mantener la remisión completa y profundizarla, destruyendo las células leucémicas residuales (enfermedad mínima residual) que en cualquier momento podrían comenzar a reproducirse y causar una recaída.
En los pacientes con leucemia mieloide aguda existen tres opciones de tratamiento de post-remisión:
- Quimioterapia de consolidación
- Quimioterapia de consolidación seguida de trasplante de médula ósea autólogo (del propio paciente – trasplante autólogo de progenitores hematopoyéticos)
- Quimioterapia de consolidación seguida de trasplante de médula ósea alogénico (de donante compatible, familiar o anónimo – trasplante alogénico de progenitores hematopoyéticos)
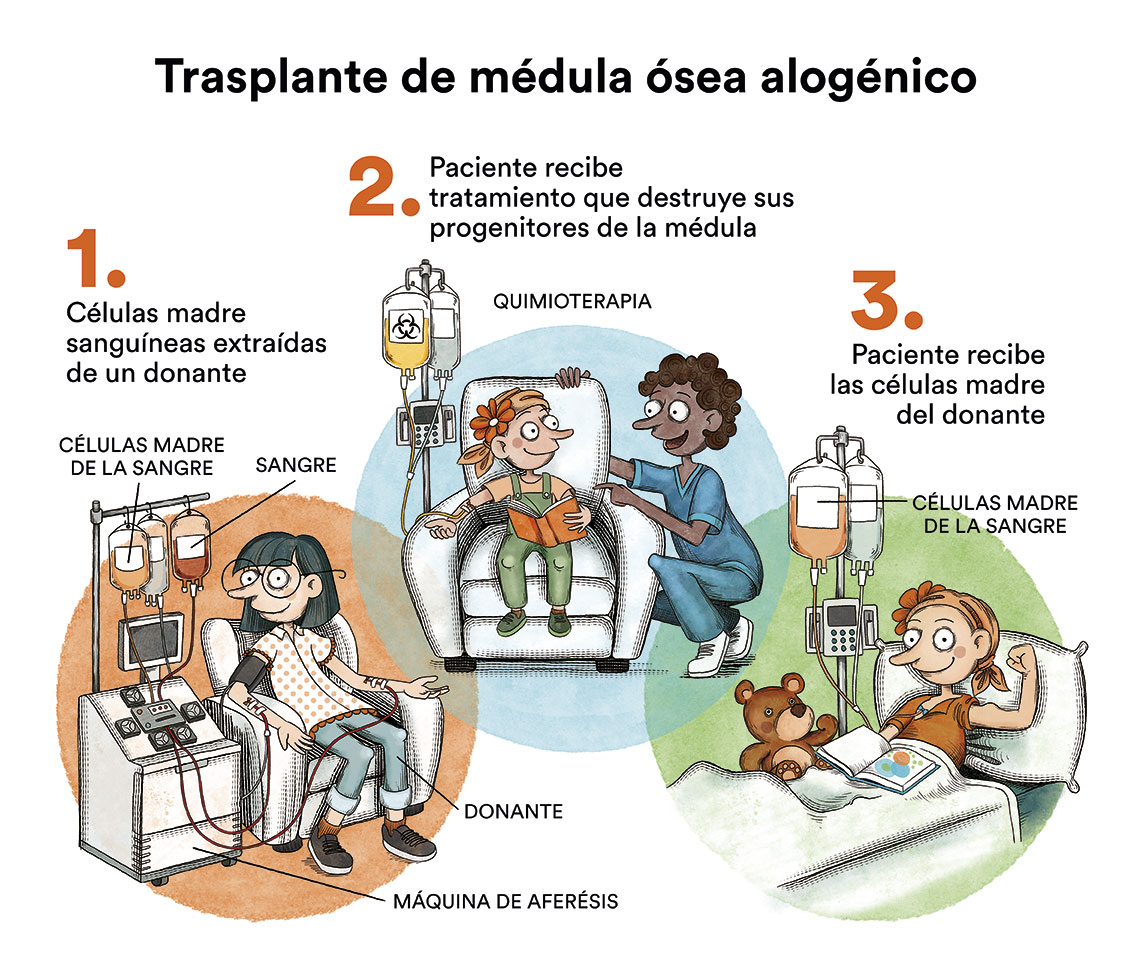
La primera opción (quimioterapia de consolidación) es la de elección en pacientes con pronóstico favorable (bajo riesgo de recidiva tras tratamiento quimioterápico intensivo) y sin enfermedad mínima residual detectable, que se estudia con pruebas de laboratorio complementarias (citometría y estudios moleculares). No hay suficientes evidencias para recomendar quimioterapia sola o quimioterapia seguida de trasplante autólogo de médula ósea, con lo que se decidirá de forma individualizada para cada paciente.
De igual modo el régimen óptimo a emplear para la consolidación y el número de ciclos de quimioterapia a administrar tampoco estén del todo establecidos. No obstante, cuando va a realizarse un trasplante suelen administrarse 1 o 2 ciclos de consolidación, mientras que cuando no se realiza trasplante se tiende a administrar un mínimo de dos ciclos.
En algunos pacientes con subtipos de leucemia mieloide aguda considerados de riesgo intermedio o alto (elevado riesgo de recaída de la enfermedad a pesar de la quimioterapia o tras una primera recaída), y con edades inferiores a 70 años y sin problemas médicos importantes adicionales, está indicada la realización de un trasplante de progenitores hematopoyéticos (médula ósea, sangre periférica o sangre de cordón umbilical) a partir de un donante compatible (trasplante alogénico), idealmente un hermano histocompatible o, en su defecto, un donante voluntario no emparentado compatible localizado a nivel mundial o una unidad de sangre de cordón umbilical. También existe la opción de realizar el trasplante con familiares 50% compatibles (haploidéntico) con resultados muy similares a los trasplantes con mayor compatibilidad.
En los pacientes mayores de 70 años, la decisión de realizar o no un trasplante alogénico se adoptar de forma individualizada. En estos casos lo importante no es la edad en sí, si no el estado general del paciente, la tolerancia a los tratamientos previos, y la disponibilidad de un buen donante. Aplica igualmente a pacientes menores de 70 años, pero con comorbilidades relevantes que no lo hacen apto para recibir un trasplante.
En los pacientes de edad avanzada y con un mal estado funcional previo, dada la esperable mala tolerancia a la quimioterapia intensiva se adoptan estrategias terapéuticas distintas. Las más empleadas son las quimioterapias a bajas dosis o los agentes hipometilantes, como la Decitabina o la Azacitidina, que tienen como finalidad retrasar al máximo la progresión de la enfermedad causando la menor toxicidad y priorizando la calidad de vida.
El tratamiento de la leucemia mieloide aguda relacionada con el tratamiento o secundaria a otras enfermedades hematológicas, no difiere demasiado del resto de leucemias mieloides agudas, aunque la probabilidad de alcanzar una remisión completa mantenida es inferior por su mayor resistencia a las quimioterapias. En estos casos, de ser factible, es habitual efectuar un trasplante alogénico de progenitores hematopoyéticos por ser la aproximación terapéutica con mayores posibilidades curativas.

Subtipos específicos: leucemia promielocítica aguda y pacientes con Síndrome de Down
Leucemia Promielocítica Aguda
Una de las leucemias que más se ha beneficiado de una estrategia terapéutica individualizada es la leucemia aguda promielocítica (LPA). En las últimas décadas, gracias a la investigación científica, se ha obtenido una mejora sustancial en su tratamiento, pasando de ser un subtipo de LMA con muy mal pronóstico a ser una enfermedad que en la mayor parte de casos responde muy bien al tratamiento. Este tipo de leucemia se caracteriza porque tiene una translocación entre los cromosomas 15 y 17 [t(15:17)], que afecta al receptor del ácido retinoico alfa (RARα o RARA) y que le confiere una alta sensibilidad al tratamiento con ácido holotransretinoico (ATRA). Es por ello que este tipo de leucemia recibe un tratamiento diferente al empleado en el resto de LMA basado en la utilización de ATRA.
La leucemia promielocítica aguda (LPA) representa el 10–15 % de todas las LMA. La edad mediana de estos pacientes es de 40 años.
El intervalo entre la aparición de los primeros síntomas y el diagnóstico de la leucemia promielocítica aguda es habitualmente inferior a dos meses debido al carácter agudo de la enfermedad. Además de síntomas atribuibles a la anemia producida por el déficit de glóbulos rojos (sensación de cansancio, debilidad, mareos, palidez), esta leucemia se caracteriza por afectar de forma muy marcada la coagulación. Por ello, las hemorragias suelen estar presentes hasta en el 75% de los pacientes en forma de hematomas en piel y mucosas; o sangrados en cualquier otra región, siendo los más peligrosos los del sistema nervioso central. De hecho, las hemorragias son responsables del 60% de las muertes en esta fase inicial de la enfermedad, con lo que es muy importante el diagnóstico rápido de la enfermedad con inicio inmediato de ATRA y factores que mejoran la coagulación. Como en el resto de leucemias, un número importante de los pacientes pueden presentar fiebre por infecciones intercurrentes secundarias a la falta de granulocitos. Es poco frecuente observar crecimiento de ganglios linfáticos, hígado o bazo.
Además de los estudios básicos en sangre y médula ósea (morfología, recuento e inmunofenotipo) a realizar en toda leucemia, en la leucemia promielocítica aguda adquieren especial relevancia la disponibilidad rápida de los resultados de citogenética y la biología molecular que confirmarán la sospecha clínica. El 80 % de los pacientes presentan la translocación t(15;17) y el 99% el gen PML-RARα, que permiten establecer un diagnóstico de seguridad con importantes implicaciones terapéuticas.
En este tipo de leucemia aguda, el tratamiento inicial también dependerá del riesgo de recaída. Para calcularlo, las clasificaciones más recientes utilizan el número de leucocitos y plaquetas en el momento del diagnóstico.
En los pacientes con riesgo favorable (leucocitos < 10000/mm3 y plaquetas > 40000/mm3), el tratamiento de inducción emplea el ATRA, perteneciente a la familia de la vitamina A, en combinación con trióxido de arsénico (ATO). El ATRA también se conoce como tretinoína (Vesanoid®), y se caracteriza por eliminar la anomalía del PML/RARα e inducir así a los promielocitos a desarrollarse en células maduras (neutrófilos).
En los pacientes de riesgo adverso (leucocitos > 10000/mm3) se asocia, además, un agente quimioterápico, habitualmente de la clase de las antraciclinas (daunorrubicina o idarubicina) con el objetivo de reducir más rápidamente el número de células de la enfermedad en la sangre.
Hay que tener presente, como efectos secundarios a este tratamiento, aparte de los comentados previamente en relación a la leucemia (infecciones, hemorragias), la posible aparición de un cuadro conocido como síndrome de diferenciación (precisamente por el efecto inmediato del ATRA en la diferenciación de los blastos) y que se caracteriza por un aumento de la cifra de leucocitos y una retención de líquidos (que se manifiesta por la aparición de infiltrados pulmonares, derrames), así como fiebre e hipotensión, y cuyo tratamiento se basa en corticoides y en la interrupción transitoria del ATRA. Por otro lado la administración de ATO puede afectar los electrolitos de la sangre y por tanto debe ir asociada a la realización de analíticas de control para la corrección de los mismos y evitar alteraciones del ritmo cardiaco.
En global, este tratamiento permite alcanzar la remisión en aproximadamente el 80-90 % de los pacientes afectos de leucemia promielocítica aguda. Tras la remisión, se continúa con los ciclos de consolidación con ATRA y ATO en número variable y, posteriormente, se realiza una fase prolongada de tratamiento de mantenimiento en la que no suelen modificarse los fármacos utilizados.
En el caso de una recidiva, el tratamiento será individualizado para cada paciente, en función de su edad, estado general y los fármacos utilizados previamente. Es en esta situación donde puede plantearse una estrategia de consolidación que incluya un trasplante alogénico de progenitores hematopoyéticos, generalmente autólogo. El trasplante alogénico en la LPA es una indicación muy infrecuente, a contemplar en caso de que fracase el trasplante autólogo, lo cual es excepcional.
Con las nuevas estrategias de tratamiento la supervivencia es muy alta siendo muy poco frecuentes las recaídas.
Pacientes con Síndrome de Down
Las personas con síndrome de Down tienen un riesgo 15 veces superior a presentar una leucemia aguda. En el caso de la leucemia mieloide aguda, la edad de presentación suele ser infantil o en adultos jóvenes.
Este grupo de pacientes presenta una elevada sensibilidad a los tratamientos quimioterápicos y esto ha posibilitado tasas de curación elevadas. Una de las principales dificultades para lograr la curación se debe a la elevada toxicidad ante algunos fármacos quimioterápicos y el elevado riesgo de infección. Es por ello por lo que diferentes grupos han conseguido aumentar la supervivencia con protocolos de tratamientos adaptados.
¿Qué probabilidades tienen de curarse los pacientes adultos de leucemia mieloide aguda?
El pronóstico de los pacientes afectados de leucemia mieloide aguda varía sustancialmente en función diversos factores que incluyen: la edad, el antecedente de tratamiento quimioterápico previo o el desarrollo de la LMA en personas con alteraciones hematológicas previas como la mielodisplasia o el síndrome mieloproliferativo, el grado de leucocitosis inicial, la presencia de determinadas anomalías citogenéticas/moleculares, así como la lentitud en la obtención de la remisión completa.
Así, los pacientes jóvenes con leucemias de riesgo bajo (grupo favorable) tienen una probabilidad de curación superior al 75%, mientras que los de riesgo intermedio que reciben un trasplante alogénico de un donante óptimo tienen una probabilidad de curación de hasta el 65-70 %. Sin embargo, un paciente con leucemias de muy alto riesgo, especialmente en caso de no alcanzar la remisión completa con el tratamiento quimioterápico, tienen muy pocas opciones de curación.
El caso concreto de la leucemia promielocítica aguda tiene una estimación del 90% de tasas de supervivencia.
Es muy importante remarcar que las estadísticas de las tasas de supervivencia en el caso de las personas con leucemia mieloide aguda son una estimación y en cada caso el equipo médico lo valorará de forma individualizada.

Seguimiento
Después de completar el tratamiento, es muy importante continuar con los controles periódicos con su equipo médico, así como con otros especialistas en caso necesario. Los controles se realizan para confirmar y ayudar en la recuperación física tras el tratamiento y hacer un seguimiento e identificación precoz de las posibles complicaciones a largo plazo. De igual manera, especialmente en los primeros meses/años tras finalizar el tratamiento, se realizarán analíticas y estudios medulares para confirmar la ausencia de reaparición de la enfermedad. Estos controles se van espaciando progresivamente. Es recomendable realizar un seguimiento como mínimo anual a largo plazo para poder detectar pronto y poder tratar, si aparecieran, las secuelas del tratamiento o de la leucemia.
Enlaces de interés sobre temas médicos relacionados con la leucemia mieloide aguda en adultos
- Tratamiento de la leucemia mieloide aguda en adultos. American Cancer Society.
- Subtipos y factores pronósticos de la leucemia mieloide aguda. American Cancer Society.
- Información sobre la leucemia promielocítica aguda. Leukemia and Lymphoma Society
- La leucemia mieloide aguda en niños y adolescentes. Leukemia and Lymphoma Society
- Guía de la leucemia mieloide aguda en adultos. American Society of Clinical Oncology
- Información sobre la leucemia mieloide aguda del adulto. AEAL
- Guía sobre la leucemia mieloide aguda para pacientes y cuidadores. Leukemia and Lymphoma Society
Enlaces de interés sobre otros temas relacionados con la leucemia linfoblástica aguda en adultos
MATERIALES TESTIMONIALES
Puedes solicitarnos los libritos en formato papel para envío gratuito en España a través del email: imparables@fcarreras.es
TRASPLANTE DE MÉDULA ÓSEA
- Guía del Trasplante de Médula Ósea. Fundación Josep Carreras
- ¿Qué es el HLA y cómo funciona? Fundación Josep Carreras
- La Enfermedad Injerto contra Receptor. Fundación Josep Carreras
- Historia del Trasplante de Médula Ósea. Fundación Josep Carreras
- ¿Cómo se realiza la búsqueda de un donante compatible anónimo? Fundación Josep Carreras
ALIMENTACIÓN
- ¿Cómo mantener una alimentación saludable durante el tratamiento? Fundación Josep Carreras
- Guía de nutrición. Leukemia & Lymphoma Society
OTROS
- Ideas sobre qué llevarme a una cámara de aislamiento. Fundación Josep Carreras contra la leucemia
- Consejos de viaje para personas con cáncer. Fundación Josep Carreras contra la leucemia
- Manual de fisioterapia en pacientes hematológicos y trasplantados. Fundación Josep Carreras contra la leucemia
- Prevención y tratamiento de la mucositis oral. Fundación Josep Carreras contra la leucemia
- La higiene bucodental en el paciente onco-hematológico. Fundación Josep Carreras contra la leucemia
- Manual fertilidad: Padecer un cáncer de la sangre y ser padre o madre. Fundación Josep Carreras contra la leucemia
- El cuidado de la piel en el paciente onco-hematológico. Fundación Josep Carreras contra la leucemia
- Manual Estética Oncológica. Fundación Josep Carreras contra la leucemia
- Leucemia y sexualidad. Fundación Josep Carreras contra la leucemia
- 7 formas de ponerse un pañuelo. Fundación Josep Carreras contra la leucemia
Enlaces de interés: entidades locales/provinciales o estatales que pueden proveerte de recursos y servicios especializados en leucemia o en pacientes oncológicos:
En España existe un gran tejido asociativo para pacientes con cáncer hematológico que, en muchos casos, puede informarte, asesorarte e incluso, realizar algunos trámites. Estos son los contactos de algunas de ellas por Comunidades Autónomas:
Todas estas organizaciones son externas a la Fundación Josep Carreras.
ESTATAL
- CEMMP (Comunidad Española de Pacientes de Mieloma Múltiple)
- AEAL (ASOCIACIÓN ESPAÑOLA DE AFECTADOS POR LINFOMA, MIELOMA y LEUCEMIA)
- AECC (ASOCIACIÓN ESPAÑOLA CONTRA EL CÁNCER). Presente en las diferentes provincias y en muchas localidades. Contactar con la sede más cercana o llamando al 900 100 036 (24h).
- AELCLES (Agrupación Española contra la Leucemia y Enfermedades de la Sangre)
- FUNDACIÓN JOSEP CARRERAS CONTRA LA LEUCEMIA
- FUNDACIÓN SANDRA IBARRA
- GEPAC (GRUPO ESPAÑOL DE PACIENTES CON CÁNCER)
- MPN España (Asociación de Afectados Por Neoplasias Mieloproliferativas Crónicas)
ANDALUCÍA
- AECC (ASOCIACIÓN ESPAÑOLA CONTRA EL CÁNCER). Presente en las diferentes provincias y en muchas localidades. Contactar con la sede más cercana.
- ALUSVI (ASOCIACIÓN LUCHA Y SONRÍE POR LA VIDA). Sevilla
- APOLEU (ASOCIACIÓN DE APOYO A PACIENTES Y FAMILIARES DE LEUCEMIA). Cádiz
ARAGÓN
- AECC (ASOCIACIÓN ESPAÑOLA CONTRA EL CÁNCER). Presente en las diferentes provincias y en muchas localidades. Contactar con la sede más cercana.
- ASPHER (ASOCIACIÓN DE PACIENTES DE ENFERMEDADES HEMATOLÓGICAS RARAS DE ARAGÓN)
- DONA MÉDULA ARAGÓN
ASTURIAS
- AECC (ASOCIACIÓN ESPAÑOLA CONTRA EL CÁNCER). Presente en las diferentes provincias y en muchas localidades. Contactar con la sede más cercana.
- ASTHEHA (ASOCIACIÓN DE TRASPLANTADOS HEMATOPOYÉTICOS Y ENFERMOS HEMATOLÓGICOS DE ASTURIAS)
CANTABRIA
- AECC (ASOCIACIÓN ESPAÑOLA CONTRA EL CÁNCER). Presente en las diferentes provincias y en muchas localidades. Contactar con la sede más cercana.
CASTILLA LA MANCHA
- AECC (ASOCIACIÓN ESPAÑOLA CONTRA EL CÁNCER). Presente en las diferentes provincias y en muchas localidades. Contactar con la sede más cercana.
CASTILLA LEÓN
- ABACES (ASOCIACIÓN BERCIANA DE AYUDA CONTRA LAS ENFERMEDADES DE LA SANGRE)
- AECC (ASOCIACIÓN ESPAÑOLA CONTRA EL CÁNCER). Presente en las diferentes provincias y en muchas localidades. Contactar con la sede más cercana.
- ALCLES (ASOCIACIÓN LEONESA CON LAS ENFERMEDADES DE LA SANGRE). León.
- ASCOL (ASOCIACIÓN CONTRA LA LEUCEMIA Y ENFERMEDADES DE LA SANGRE). Salamanca.
CATALUÑA
- ASSOCIACIÓ FÈNIX. Solsona
- FECEC (FEDERACIÓ CATALANA D’ENTITATS CONTRA EL CÁNCER
- FUNDACIÓ KÁLIDA. Barcelona
- FUNDACIÓ ROSES CONTRA EL CÀNCER. Roses
- LLIGA CONTRA EL CÀNCER COMARQUES DE TARRAGONA I TERRES DE L’EBRE. Tarragona
- MielomaCAT
- ONCOLLIGA BARCELONA. Barcelona
- ONCOLLIGA GIRONA. Girona
- ONCOLLIGA COMARQUES DE LLEIDA. Lleida
- ONCOVALLÈS. Vallès Oriental
- OSONA CONTRA EL CÀNCER. Osona
- SUPORT I COMPANYIA. Barcelona
- VILASSAR DE DALT CONTRA EL CÀNCER. Vilassar de Dalt
COMUNIDAD VALENCIANA
- AECC (ASOCIACIÓN ESPAÑOLA CONTRA EL CÁNCER). Presente en las diferentes provincias y en muchas localidades. Contactar con la sede más cercana.
- ASLEUVAL (ASOCIACIÓN DE PACIENTES DE LEUCEMIA, LINFOMA, MIELOMA Y OTRAS ENFERMEDADES DE LA SANGRE DE VALENCIA)
EXTREMADURA
- AECC (ASOCIACIÓN ESPAÑOLA CONTRA EL CÁNCER). Presente en las diferentes provincias y en muchas localidades. Contactar con la sede más cercana.
- AFAL (AYUDA A FAMILIAS AFECTADAS DE LEUCEMIAS, LINFOMAS; MIELOMAS Y APLASIAS)
- AOEX (ASOCIACIÓN ONCOLÓGICA EXTREMEÑA)
GALICIA
- AECC (ASOCIACIÓN ESPAÑOLA CONTRA EL CÁNCER). Presente en las diferentes provincias y en muchas localidades. Contactar con la sede más cercana.
- ASOTRAME (ASOCIACIÓN GALLEGA DE AFECTADOS POR TRASPLANTES MEDULARES)
ISLAS BALEARES
- ADAA (ASSOCIACIÓ D’AJUDA A L’ACOMPANYAMENT DEL MALALT DE LES ILLES BALEARS)
- AECC (ASOCIACIÓN ESPAÑOLA CONTRA EL CÁNCER). Presente en las diferentes provincias y en muchas localidades. Contactar con la sede más cercana.
ISLAS CANARIAS
- AECC (ASOCIACIÓN ESPAÑOLA CONTRA EL CÁNCER). Presente en las diferentes provincias y en muchas localidades. Contactar con la sede más cercana.
- AFOL (ASOCIACIÓN DE FAMILIAS ONCOHEMATOLÓGICAS DE LANZAROTE)
- FUNDACIÓN ALEJANDRO DA SILVA
LA RIOJA
- AECC (ASOCIACIÓN ESPAÑOLA CONTRA EL CÁNCER). Presente en las diferentes provincias y en muchas localidades. Contactar con la sede más cercana.
MADRID
- AECC (ASOCIACIÓN ESPAÑOLA CONTRA EL CÁNCER). Presente en las diferentes provincias y en muchas localidades. Contactar con la sede más cercana.
- AEAL (ASOCIACIÓN ESPAÑOLA DE LEUCEMIA Y LINFOMA)
- CRIS CONTRA EL CÁNCER
- FUNDACIÓN LEUCEMIA Y LINFOMA
MURCIA
- AECC (ASOCIACIÓN ESPAÑOLA CONTRA EL CÁNCER). Presente en las diferentes provincias y en muchas localidades. Contactar con la sede más cercana.
NAVARRA
- AECC (ASOCIACIÓN ESPAÑOLA CONTRA EL CÁNCER). Presente en las diferentes provincias y en muchas localidades. Contactar con la sede más cercana.
PAÍS VASCO
- AECC (ASOCIACIÓN ESPAÑOLA CONTRA EL CÁNCER). Presente en las diferentes provincias y en muchas localidades. Contactar con la sede más cercana.
- PAUSOZ-PAUSO. Bilbao
CIUDADES AUTÓNOMAS DE CEUTA Y MELILLAS
- AECC CEUTA (ASOCIACIÓN ESPAÑOLA CONTRA EL CÁNCER)
- AECC MELILLA (ASOCIACIÓN ESPAÑOLA CONTRA EL CÁNCER)
Apoyo y ayuda
Te invitamos también a seguirnos a través de nuestras redes sociales principales (Facebook, Twitter e Instagram) en las que, a menudo, compartimos testimonios de superación.
Si resides en España, también puedes ponerte en contacto con nosotros enviándonos un correo electrónico a imparables@fcarreras.es para que te ayudemos a ponerte en contacto con otras familias que han superado esta enfermedad.